Prognose der Verkehrslage in der Region Hannover
Die primäre Anforderung der Verkehrsteilnehmer im Bereich des Straßenverkehrs ist die Kenntnis der aktuellen Verkehrslage. Diese basiert in der Regel auf der wirklich benötigten Reisezeit von sehr vielen Verkehrsteilnehmern, deren Daten häufig im Kontext von Routingdiensten abgegriffen werden.
Im Rahmen von Data4UrbanMobility wurden Werkzeuge entwickelt um eine ganglineinbasierte Prognose der Verkehrslage zu ermöglichen. Die folgende Abbildung zeigt eine Oberfläche auf der typische Ganglinienverläufe und Ausreißer visualisiert werden.
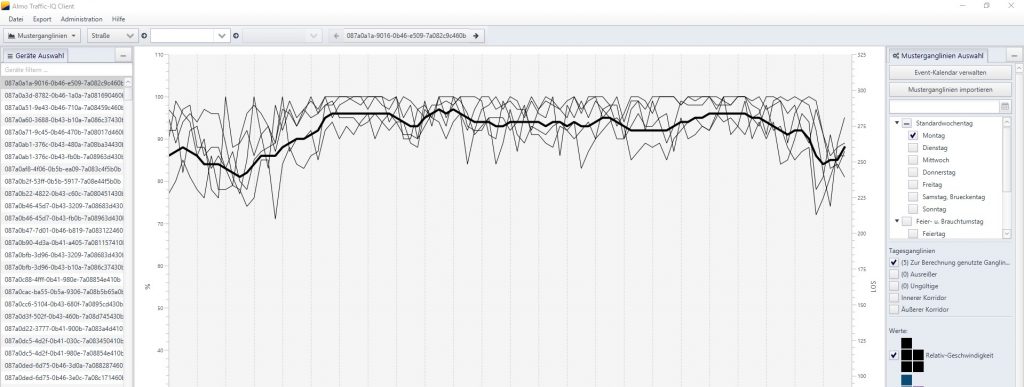
Die Prognose der Verkehrslage kann dann mittels einer Karte für den Endnutzer visualisert werden:
Erste Version der MIC-App bereitgestellt
Eine erste Version der MIC-App (Move in the City) konnte allen Partnerinnen und Partnern des Projekts und einer geschützten Nutzer*innengruppe der Öffentlichkeit zur Verfügung gestellt werden. Die mobile App MiC ist ein Instrument zur Datenerhebung.
Dabei verknüpft MiC – eine Entwicklung des Institute for Sustainable Urbanism ISU der TU Braunschweig und Projektionisten GmbH Hannover – das wachsende Bewusstsein und die Notwendigkeit für digitale Bürger*innenrechte mit den Potentialen mittels der Auswertung großer Datenmengen neue Formen der menschzentrierten Entwicklung von Stadt und Mobilität zu ermöglichen stellt eine Möglichkeit dar, sich aktiv als Bürgerwissenschaftlerin und Bürgerwissenschaftler an der Forschung und Entwicklung der Mobilität für alle in der Stadt der Zukunft zu beteiligen.
MiC erhebt – durch die Nutzerinnen und Nutzer gesteuert – Daten zu Strecken und Art der Fortbewegung. Diese Daten werden pseudonymisiert, so dass ein Rückschluss auf die jeweilige Person nicht mehr möglich ist. Wichtig ist die Vielzahl der Nutzerinnen und Nutzer – nicht die einzelne Bewegung. Die Stadt der Zukunft zeichnet sich aus durch den barrierearmen Zugang zu Mobilität und Erreichbarkeit für alle. Der holistische Ansatz der Forscherinnen und Forscher des Institute for Sustainable Urbanism ISU (TU Braunschweig) sowie der Projektbeteiligten betrachtet Stadt dabei auf verschiedenen Maßstabsebenen und bringt intelligente Planungen – wie z.B. die 5-Minuten Stadt –, Städtebau und innovative Technologien zusammen. Für ein umfassendes Verständnis individueller Mobilität und darauf aufbauende neue Methoden und Werkzeuge für integrierte Verkehrs- und Stadtplanung werden mittels der MiC-App uns umfangreiche und detaillierte Daten darüber geliefert, wie und auf welchem Wege wir uns in der Stadt fortbewegen.
Entwicklungsstand:
In der ersten Version ermöglicht das Stadtforschungstool MiC den Nutzer*innen durch eine einfach Handhabung das Starten und Beenden der „Tracking-Time“ (Bild 1). Wichtig ist, die Nutzer*innen entscheidet selber über den Zeitraum. Als erstes Ergebnis für die Nutzer*innen steht eine Zusammenfassung ihrer bisher aufgezeichneten Routen (Bild2). In den Einstellung (Bild 3) kann der Nutzer sich aktiv an Feedback beteiligen (Bild 4) sowie seinen Account und somit seiner zur Verfügung gestellten Daten löschen (Bild 5).
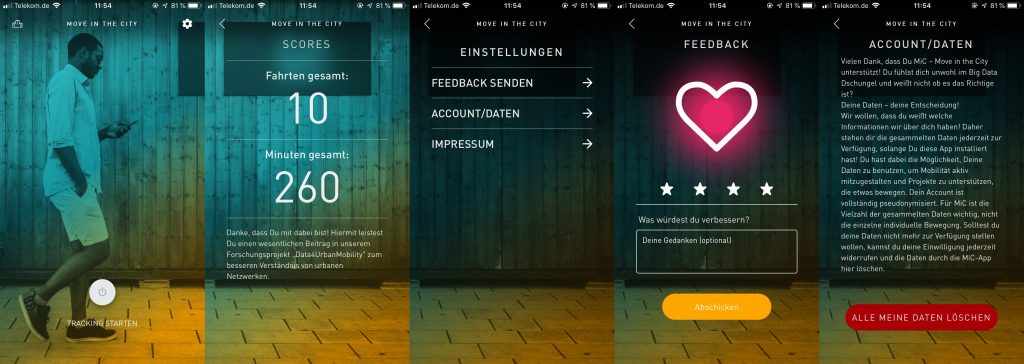
von links nach recht: Bild1-5 MIC App Interface – Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0)
Die aktuelle Weiterentwicklung sieht eine Visualisierung der Routen für den jeweiligen Nutzer vor.
Um Teil der Testgruppe zu werden ist zur Zeit noch eine Anmeldung unter: www.mic-app.org notwendig. Die Anwendung ist nicht frei im App Store / GooglePlay Store zu erhalten.
Auf der Internetseite www.mic-app.org wird zusätzlich detailliert auf häufige Fragen (FAQ) zur Anwendung sowie über Entwicklungen und Neuheiten informiert
D4UM Plattform und Dashboard V2
Die neue Version der Plattform inklusive des Dashboards gibt noch detailliertere Auskünfte über die Verkehrssituation
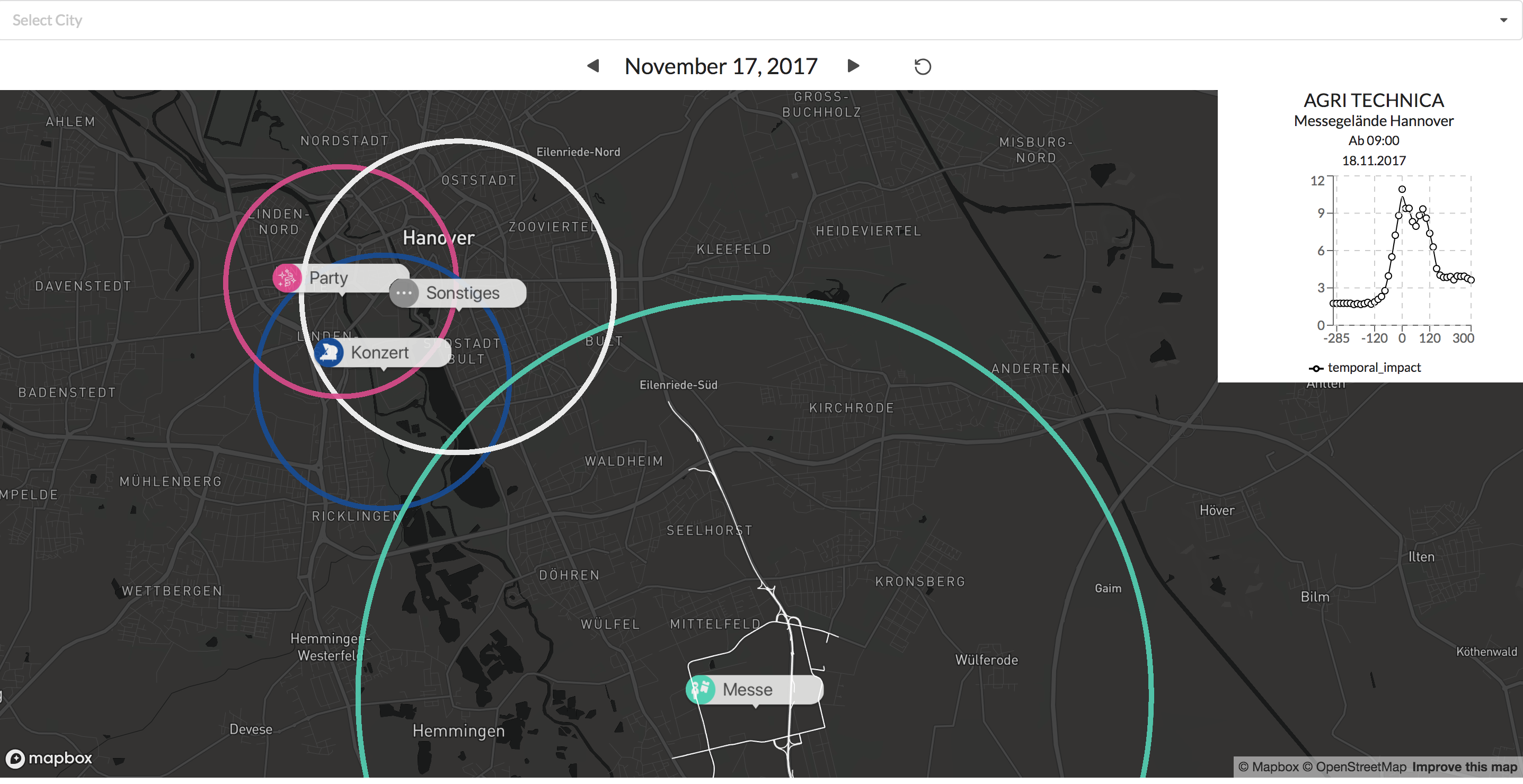
Die farblich unterschiedlichen Label lassen eine schnelle Unterscheidung zwischen den verschiedenen Event typen zu. Durch das klicken auf eines der Events wird der typically affected subgraph angezeigt für diesen Eventtyp.
Beispiele: Visualisierungen eines Konzerts und eines Fußballspiels
Zusätzlich gibt der Graph in der oberen rechten Ecke Auskunft über die Verkehrssituation vor und nach dem Eventstart.
{API}
Es wurden die API Endpunkte mit zusätzlichen Information erweitert.
Diese werden mittels der als Teil der Forschung entwickelten Modellen erstellt.
Erste Version der D4UM-App bereitgestellt
Eine erste Version der D4UM-App konnte allen Partnern des Projekts zur Verfügung gestellt werden. Die App stellt eine Möglichkeit dar, sich Fahrtauskünfte mit dem öffentlichen Personennahverkehr in Niedersachsen und Bremen (Datengrundlage: EFA – elektronische Fahrplanauskunft für Niedersachsen und Bremen) ausgeben zu lassen. Im Fokus stand hierbei, dass der Nutzer schnell und einfach an die für ihn wichtigen Informationen gelangen kann, um so seine Reise möglichst simpel planen zu können.
Folgende Funktionen dienen dabei in der ersten Version der schnellen Auskunft:
Abfahrten und Verbindungen
Über die Funktion Abfahrten lassen sich Abfahrtszeiten an einer bestimmten oder an nahegelegenen Haltestellen ermitteln. Unter Verbindungen können hingegen Fahrtvorschläge von einem Startpunkt (Adresse oder Haltestelle) zu einem Zielpunkt gesucht werden. Zeiten stehen dabei auch in Echtzeit zur Verfügung, sodass auch Verspätungen direkt von dem Nutzer erkannt werden können.
Karte
Über die Karte sind alle Haltestellen zu finden, sodass sich der Nutzer einen Überblick über die nähere Umgebung oder auch den Weg zur Haltestelle oder einem Ziel verschaffen kann.
Wird auf der Karte auf ein Haltestellensymbol oder den zugehörigen Haltestellennamen geklickt, öffnet sich der Abfahrtsmonitor zu dieser Haltestelle. Die nächsten Abfahrten können somit auch über diesen Weg aufgerufen werden.
Darüber hinaus kann sich der Nutzer auch den Verlauf seiner Fahrt anzeigen lassen.
Menü/Einstellungen
Weitere Funktionen und Einstellungen finden sich ergänzend im Menü der App.
Der Nutzer bekommt hier zum einen die Möglichkeit, dass erweiterte Einstellungen zu den Suchanfragen bei Verbindungen oder Abfahrten vorgenommen werden können, und zum anderen, dass er weitere Features verwenden kann. Darunter befindet sich zum Beispiel das Feedbackformular. Hierüber kann unkompliziert Kontakt mit den Entwicklern der D4UM-App per Mail aufgenommen werden. Icons ermöglichen es, dass ein Eindruck zu der App übermittelt werden kann. Ein weiteres Feld für Freitext bietet zudem Platz für individuelle Kritik und einer Meinung zu der App. So kann in Zukunft kundennah an der App weiterentwickelt und einfach auf Wünsche und Meinungen reagiert werden.
Quantifizierungen und Vorhersage von Auswirkungen von Veranstaltungen
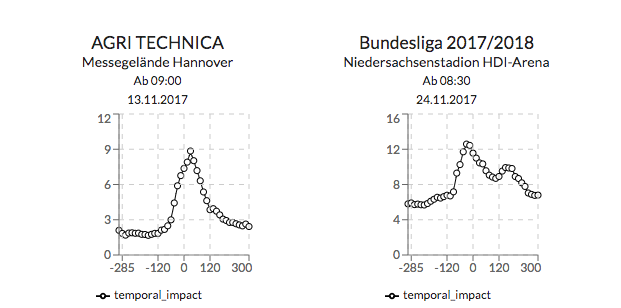
Neue Data4UrbanMobility-Forschungsergebnisse ermöglichen es, die räumlichen Auswirkungen von Veranstaltungen zu quantifizieren und vorherzusagen. Dazu werden zusammenhängende, betroffene Straßenabschnitte in der Nähe von Veranstaltungen identifiziert. Auf dieser Grundlage kann dann die räumliche Auswirkung quantifiziert werden. Das Verfahren ist in der folgenden Grafik dargestellt.
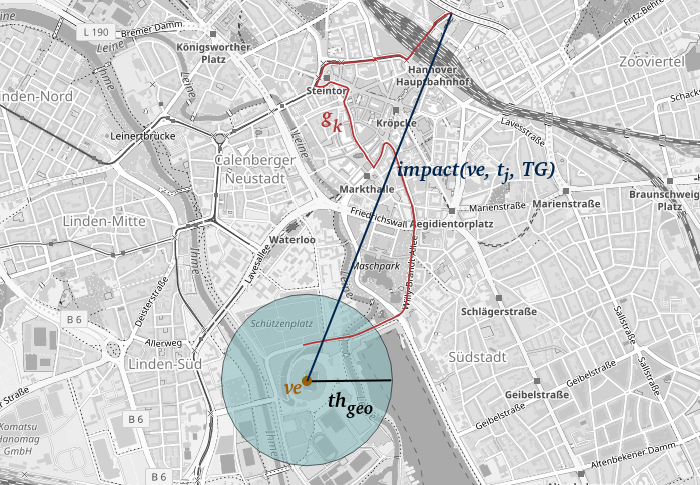
Hier in Gelb markiert ist eine Veranstaltung, in Rot betroffene Straßenabschnitte und in Dunkelblau die gemessene Auswirkung. Weiterhin wurden Verfahren des Maschinellen Lernens angewandt, um diese Auswirkungen zu prognostizieren. Dabei konnte der Fehler gegenüber bestehenden state-of-the-art Ansätzen um bis zu 40% verringert werden.
D4UM – Plattform V1 fertiggestellt
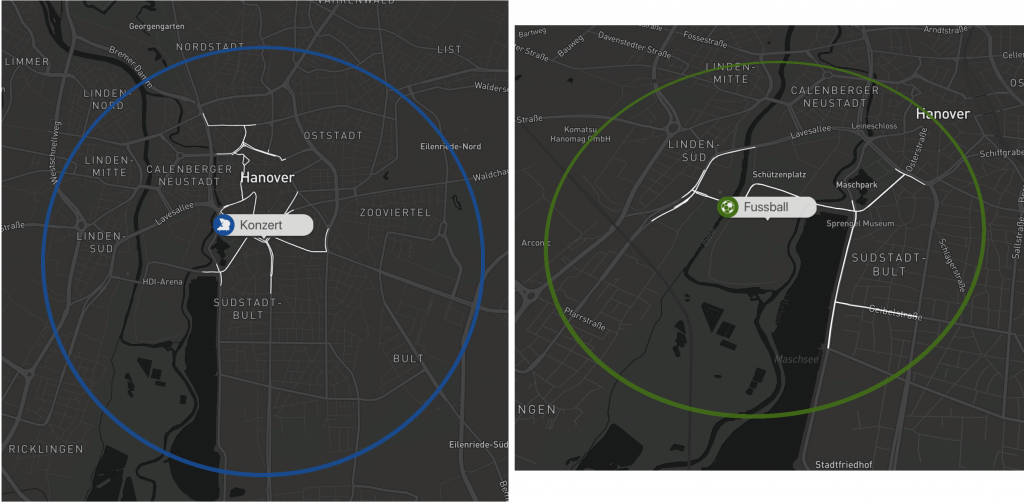
Die erste Version der Data4UrbanMobility Plattform wurde fertiggestellt. Dazu wurde zunächst eine 3-Schichten Architektur der Plattform konzipiert und implementiert. Die Plattform bietet RESTfull Webservices für Mobilitätsapplikationen wie Dashboard-Anwendungen oder Apps an. Als erste Beispielanwendung wurde dazu eine interaktive Karte entwickelt, die die Auswirkungen von Veranstaltungen visualisiert. Ein Ausschnitt aus der Anwendung ist im folgenden Screenshot zu sehen.
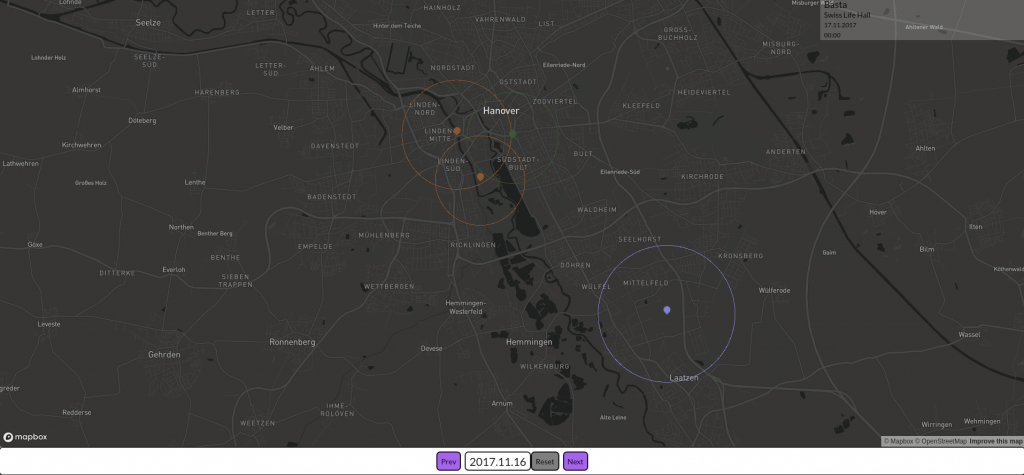
Zu sehen sind 4 Veranstaltungen in Hannover. Die Farben entsprechen dabei unterschiedlichen Veranstaltungsarten (etwa Konzerte, Messen, Fußballspiele). Die Kreise visualisieren die räumlichen Auswirkungen, die diese Veranstaltungen auf den Verkehr hatten.
Umfangreicher Anforderungskatalog
Die Data4UrbanMobility Anforderungsanalyse umfasst die Erfassung der Anforderungen der Anwendungspartner Region Hannover (RH) und Wolfsburg AG (WAG), sowie der nicht-funktionalen Anforderungen. Aus den Anforderungen der AnwendungspartnerInnen (RH und WAG), die von MOMA erhoben wurden, sind von L3S Forschungsfragen für die Datenanalyse abgeleitet worden, die sich speziell auf die Informationsbedürfnisse der AnwenderInnen beziehen und im weiteren Projektverlauf adressiert werden.
Die aktuelle Forschungsfragen adressieren insbesondere:
- Automatische Verifikation von Verkehrswarnmeldungen und Prognose von deren Auswirkungen.
- Identifikation von Veranstaltungen und Prognose verkehrsrelevanter Auswirkungen.
- Korrelation von IV-Reiseflussdaten, EFA-Querylogs, Warnmeldungen und Twitterfeeds.
- Bestimmung von optimalen Reisezeitpunkte.
Wachsende Datensammlung
Das ISU hat einen umfassende Datenmatrix mit potentiellen Quellen für mobilitätsrelevante Daten erstellt. Das von L3S entwickelte Data4UrbanMobility Datenmodell beschreibt alle projektrelevanten Daten und setzt diese in Verbindung um die Daten sowohl für die Analyse als auch für die Anwendungen und Apps einheitlich zur Verfügung zu stellen. Die ausgewählten Datenquellen sind von L3S in das Data4UrbanMobility Datenmodell überführt. Einige der Datenquellen wie EFA-logs, und IV-Daten sind dabei auf deren Qualität geprüft worden.
Um die Datenintegration zu ermöglichen sind Werkzeuge zur Extraktion der relevanten Daten aus Mobilitätsrelevanten Datenquellen entwickelt worden:
- Straßen- und Graphextraktion aus OpenStreetMap
- EFA-Anfragen Bulkloader für die Extraktion der ÖPNV Anfragen aus EFA Logs
- Integration von Daten aus dem Zentralen Haltestellen Verzeichnis (ZHV) inklusive Verknüpfung der Daten mit den EFA-Anfragen
Die aktuelle Datensammlung (Stand: 12 Dezember 2017) umfasst:
EFA-Logs: 17 Mio. Suchanfragen
IV-Daten: 174 Tsd. Straßen, alle 15 Minuten
GTFS-Daten: 90 Tsd. Haltestellen, 2,6 Tsd. Routen
Wetter: Radolan Regenraster
Twitter: 2,5 Mio. Tweets ab Juni 2017
OSM: 440 Tsd. Straßen
Events: 21 Tsd. Veranstaltungen (14.08.2016-17.07.2018)
Warnmeldungen: 13 Tsd. Warnmeldungen (ab 06.2017)
Visualisierungen der ÖPNV Informationen
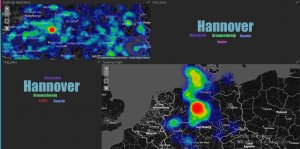
Zur intuitiven Analyse von mobilitätsrelevanten Informationen, insbesondere von ÖPNV Informationen, wurde von den PROJEKTIONISTEN (PROJ) eine Dashboard-Webapplikation konzipiert. Erste Prototypen visualisieren Anfragen an das regionale Fahrplanauskunftsystem EFA (www.efa.de) und dienen als Ausgangsbasis für explorative Analysen und die Implementierung der produktiven Version des Dashboards. Im Folgenden ist eine im Dashboard integrierte Visualisierung der häufigsten Start- und Ziel-punkte zu sehen.
Analysen der EFA-Logs
Als erste Forschungsfrage wird aktuell die Analyse der Auswirkungen der Veranstaltungen auf dem ÖPNV mit Methoden des Maschinellen Lernens analysiert. Hierzu wurden in explorativen Datenanalysen der Einfluss von großen Veranstaltungen wie z.B. Fussballspielen und mittelgroßen Veranstaltungen, etwa Konzerte, auf Anfragen an den ÖPNV betrachtet. Als Grundlage für umfassende Analysen wurden mit Hilfe visueller Methoden exemplarisch Korrelation zwischen ÖPNV-Nachfrage und Veranstaltungszeiträumen detektiert.
Dabei zeichnen sich z.B. für Hannovers Innenstadt klare, sternförmige Muster ab, die zentrale Mobilitätsknoten identifizieren.
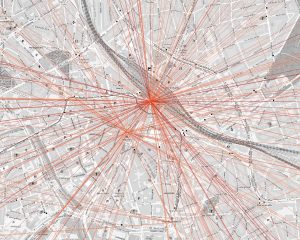
Das Bild stellt die Luftlinie zwischen Start- und Ziel-Ort der Anfragen dar. Dabei entsprechen dunklere Farben häufigeren Strecken. Hier werden deutlich Hannover Hauptbahnhof und Hannover Kröpcke (die zentrale U-Bahn Station) als Mobilitätsknoten identifiziert.
Analysen der Nachfrage für einzelne Stationen lassen wochentagspezifische Muster erkennen.
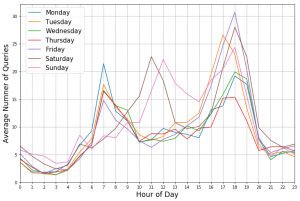
Hier dargestellt sind die durchschnittliche Anzahl der Anfragen mit der Ziel-Haltestelle “Hannover Stadionbrücke”. Zu erkennen sind vor allem Unterschiede zwischen Werktagen und dem Wochenende.
Auch der Einfluss von Veranstaltungen kann mit Hilfe der Anfragen visualisiert werden:
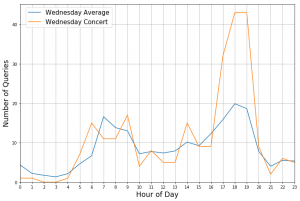
Dargestellt sind die Anzahl der Anfragen mit Ziel “Hannover Stadionbrücke” für Mittwoch, den 26.04.2017 (Orange) sowie die durchschnittlichen Anzahl von Anfragen, die mittwochs mit gleichem Ziel gestellt wird (Blau).
An diesem Tag fand in einer nahe gelegenen Konzerthalle ein Konzert statt, das um 20 Uhr begann. Die signifikante Abweichung zwischen 17 und 19 Uhr wurde sehr wahrscheinlich von den anreisenden Gästen verursacht wurde. Dies illustriert, dass Anfragen an den ÖPNV eine wertvolle Informationsquelle sein können, um Prognosen über die Auswirkung von Veranstaltungen auf Mobilität zu erstellen.
Cardiac function in a large animal model of myocardial infarction at 7 T: deep learning based automatic segmentation increases reproducibility. Kollmann, Alena; Lohr, David; Ankenbrand, Markus J.; Bille, Maya; Terekhov, Maxim; Hock, Michael; Elabyad, Ibrahim; Baltes, Steffen; Reiter, Theresa; Schnitter, Florian; Bauer, Wolfgang R.; Hofmann, Ulrich; Schreiber, Laura M. (2024). 14(1) 11009.
Cardiac magnetic resonance (CMR) imaging allows precise non-invasive quantification of cardiac function. It requires reliable image segmentation for myocardial tissue. Clinically used software usually offers automatic approaches for this step. These are, however, designed for segmentation of human images obtained at clinical field strengths. They reach their limits when applied to preclinical data and ultrahigh field strength (such as CMR of pigs at 7 T). In our study, eleven animals (seven with myocardial infarction) underwent four CMR scans each. Short-axis cine stacks were acquired and used for functional cardiac analysis. End-systolic and end-diastolic images were labelled manually by two observers and inter- and intra-observer variability were assessed. Aiming to make the functional analysis faster and more reproducible, an established deep learning (DL) model for myocardial segmentation in humans was re-trained using our preclinical 7 T data (n\thinspace=\thinspace772 images and labels). We then tested the model on nthinspace=thinspace288 images. Excellent agreement in parameters of cardiac function was found between manual and DL segmentation: For ejection fraction (EF) we achieved a Pearson's r of 0.95, an Intraclass correlation coefficient (ICC) of 0.97, and a Coefficient of variability (CoV) of 6.6%. Dice scores were 0.88 for the left ventricle and 0.84 for the myocardium.
Ageing-Dependent Thiol Oxidation Reveals Early Oxidation of Proteins with Core Proteostasis Functions. Jonak, Katarzyna; Suppanz, Ida; Bender, Julian; Chacinska, Agnieszka; Warscheid, Bettina; Topf, Ulrike (2024). 7(5)
Oxidative post-translational modifications of protein thiols are well recognized as a readily occurring alteration of proteins, which can modify their function and thus control cellular processes. The development of techniques enabling the site-specific assessment of protein thiol oxidation on a proteome-wide scale significantly expanded the number of known oxidation-sensitive protein thiols. However, lacking behind are large-scale data on the redox state of proteins during ageing, a physiological process accompanied by increased levels of endogenous oxidants. Here, we present the landscape of protein thiol oxidation in chronologically aged wild-type Saccharomyces cerevisiae in a time-dependent manner. Our data determine early-oxidation targets in key biological processes governing the de novo production of proteins, protein folding, and degradation, and indicate a hierarchy of cellular responses affected by a reversible redox modification. Comparison with existing datasets in yeast, nematode, fruit fly, and mouse reveals the evolutionary conservation of these oxidation targets. To facilitate accessibility, we integrated the cross-species comparison into the newly developed OxiAge Database.
Magnetism in the axion insulator candidate Eu\($_\mathbf5$\)In\($_\mathbf2$\)Sb\($_\mathbf6$\). Rahn, M. C.; Wilson, M. N.; Hicken, T. J.; Pratt, F. L.; Wang, C.; Orlandi, F.; Khalyavin, D. D.; Manuel, P.; Veiga, L. S. I.; Bombardi, A.; Francoual, S.; Bereciartua, P.; Sukhanov, A. S.; Thompson, J. D.; Thomas, S. M.; Rosa, P. F. S.; Lancaster, T.; Ronning, F.; Janoschek, M. (2024). 109(17) 174404.
\($\mathrmEu_5\mathrmIn_2\mathrmSb_6$\) is a member of a family of orthorhombic nonsymmorphic rare-earth intermetallics that combines large localized magnetic moments and itinerant exchange with a low carrier density and perpendicular glide planes. This may result in special topological crystalline (wallpaper fermion) or axion insulating phases. Recent studies of \($\mathrmEu_5\mathrmIn_2\mathrmSb_6$\) single crystals have revealed colossal negative magnetoresistance and multiple magnetic phase transitions. Here, we clarify this ordering process using neutron scattering, resonant elastic x-ray scattering, muon spin-rotation, and magnetometry. The nonsymmorphic and multisite character of \($\mathrmEu_5\mathrmIn_2\mathrmSb_6$\) results in coplanar noncollinear magnetic structures with an Ising-like net magnetization along the \($a$\) axis. A reordering transition, attributable to competing ferro- and antiferromagnetic couplings, manifests as the onset of a second commensurate Fourier component. In the absence of spatially resolved probes, the experimental evidence for this low-temperature state can be interpreted either as an unusual double-\($q$\) structure or in a phase separation scenario. The net magnetization produces variable anisotropic hysteretic effects which also couple to charge transport. The implied potential for functional domain physics and topological transport suggests that this structural family may be a promising platform to implement concepts of topological antiferromagnetic spintronics.
Intermembrane Space-Localized TbTim15 Is an Essential Subunit of the Single Mitochondrial Inner Membrane Protein Translocase of Trypanosomes. von Känel, Corinne; Oeljeklaus, Silke; Wenger, Christoph; Stettler, Philip; Harsman, Anke; Warscheid, Bettina; Schneider, André (2024).
All mitochondria import \($>$\)95% of their proteins from the cytosol. This process is mediated by protein translocases in the mitochondrial membranes, whose subunits are generally highly conserved. Most eukaryotes have two inner membrane protein translocases (TIMs) that are specialized to import either presequence-containing or mitochondrial carrier proteins. In contrast, the parasitic protozoan Trypanosoma brucei has a single TIM complex consisting of one conserved and five unique subunits. Here, we identify candidates for new subunits of the TIM or the presequence translocase-associated motor (PAM) using a protein-protein interaction network of previously characterized TIM and PAM subunits. This analysis reveals that the trypanosomal TIM complex contains an additional trypanosomatid-specific subunit, designated TbTim15. TbTim15 is associated with the TIM complex, lacks transmembrane domains, and localizes to the intermembrane space. TbTim15 is essential for procyclic and bloodstream forms of trypanosomes. It contains two twin CX(9)C motifs and mediates import of both presequence-containing and mitochondrial carrier proteins. While the precise function of TbTim15 in mitochondrial protein import is unknown, our results are consistent with the notion that it may function as an import receptor for the non-canonical trypanosomal TIM complex.
Identification of TFG- and Autophagy-Regulated Proteins and Glycerophospholipids in B Cells. Steinmetz, Tobit D.; Thomas, Jana; Reimann, Lena; Himmelreich, Ann-Kathrin; Schulz, Sebastian R.; Golombek, Florian; Castiglione, Kathrin; Jäck, Hans-Martin; Brodesser, Susanne; Warscheid, Bettina; Mielenz, Dirk (2024). 23(5) 1615–1633.
Autophagy supervises the proteostasis and survival of B lymphocytic cells. Trk-fused gene (TFG) promotes autophagosome-lysosome flux in murine CH12 B cells, as well as their survival. Hence, quantitative proteomics of CH12tfgKO and WT B cells in combination with lysosomal inhibition should identify proteins that are prone to lysosomal degradation and contribute to autophagy and B cell survival. Lysosome inhibition via NH(4)Cl unexpectedly reduced a number of proteins but increased a large cluster of translational, ribosomal, and mitochondrial proteins, independent of TFG. Hence, we propose a role for lysosomes in ribophagy in B cells. TFG-regulated proteins include CD74, BCL10, or the immunoglobulin JCHAIN. Gene ontology (GO) analysis reveals that proteins regulated by TFG alone, or in concert with lysosomes, localize to mitochondria and membrane-bound organelles. Likewise, TFG regulates the abundance of metabolic enzymes, such as ALDOC and the fatty acid-activating enzyme ACOT9. To test consequently for a function of TFG in lipid metabolism, we performed shotgun lipidomics of glycerophospholipids. Total phosphatidylglycerol is more abundant in CH12tfgKO B cells. Several glycerophospholipid species with similar acyl side chains, such as 36:2 phosphatidylethanolamine and 36:2 phosphatidylinositol, show a dysequilibrium. We suggest a role for TFG in lipid homeostasis, mitochondrial functions, translation, and metabolism in B cells.
The Mba1 Homologue of Trypanosoma Brucei Is Involved in the Biogenesis of Oxidative Phosphorylation Complexes. Wenger, Christoph; Harsman, Anke; Niemann, Moritz; Oeljeklaus, Silke; von Känel, Corinne; Calderaro, Salvatore; Warscheid, Bettina; Schneider, André (2023). 119(5) 537–550.
Consistent with other eukaryotes, the Trypanosoma brucei mitochondrial genome encodes mainly hydrophobic core subunits of the oxidative phosphorylation system. These proteins must be co-translationally inserted into the inner mitochondrial membrane and are synthesized by the highly unique trypanosomal mitoribosomes, which have a much higher protein to RNA ratio than any other ribosome. Here, we show that the trypanosomal orthologue of the mitoribosome receptor Mba1 (TbMba1) is essential for normal growth of procyclic trypanosomes but redundant in the bloodstream form, which lacks an oxidative phosphorylation system. Proteomic analyses of TbMba1-depleted mitochondria from procyclic cells revealed reduced levels of many components of the oxidative phosphorylation system, most of which belong to the cytochrome c oxidase (Cox) complex, three subunits of which are mitochondrially encoded. However, the integrity of the mitoribosome and its interaction with the inner membrane were not affected. Pull-down experiments showed that TbMba1 forms a dynamic interaction network that includes the trypanosomal Mdm38/Letm1 orthologue and a trypanosome-specific factor that stabilizes the CoxI and CoxII mRNAs. In summary, our study suggests that the function of Mba1 in the biogenesis of membrane subunits of OXPHOS complexes is conserved among yeast, mammals and trypanosomes, which belong to two eukaryotic supergroups.
A Msp1-containing Complex Removes Orphaned Proteins in the Mitochondrial Outer Membrane of T. Brucei. Gerber, Markus; Suppanz, Ida; Oeljeklaus, Silke; Niemann, Moritz; Käser, Sandro; Warscheid, Bettina; Schneider, André; Dewar, Caroline E. (2023). 6(11)
The AAA-ATPase Msp1 extracts mislocalised outer membrane proteins and thus contributes to mitochondrial proteostasis. Using pulldown experiments, we show that trypanosomal Msp1 localises to both glycosomes and the mitochondrial outer membrane, where it forms a complex with four outer membrane proteins. The trypanosome-specific pATOM36 mediates complex assembly of \($\alpha$\)-helically anchored mitochondrial outer membrane proteins such as protein translocase subunits. Inhibition of their assembly triggers a pathway that results in the proteasomal digestion of unassembled substrates. Using inducible single, double, and triple RNAi cell lines combined with proteomic analyses, we demonstrate that not only Msp1 but also the trypanosomal homolog of the AAA-ATPase VCP are implicated in this quality control pathway. Moreover, in the absence of VCP three out of the four Msp1-interacting mitochondrial proteins are required for efficient proteasomal digestion of pATOM36 substrates, suggesting they act in concert with Msp1. pATOM36 is a functional analog of the yeast mitochondrial import complex complex and possibly of human mitochondrial animal-specific carrier homolog 2, suggesting that similar mitochondrial quality control pathways linked to Msp1 might also exist in yeast and humans.
Conformational Regulation and Target-Myristoyl Switch of Calcineurin B Homologous Protein 3. Becker, Florian; Fuchs, Simon; Refisch, Lukas; Drepper, Friedel; Bildl, Wolfgang; Schulte, Uwe; Liang, Shuo; Heinicke, Jonas Immanuel; Hansen, Sierra C.; Kreutz, Clemens; Warscheid, Bettina; Fakler, Bernd; Mymrikov, Evgeny V.; Hunte, Carola (2023). 12
Calcineurin B homologous protein 3 (CHP3) is an EF-hand Ca(2+)-binding protein involved in regulation of cancerogenesis, cardiac hypertrophy, and neuronal development through interactions with sodium/proton exchangers (NHEs) and signalling proteins. While the importance of Ca(2+) binding and myristoylation for CHP3 function has been recognized, the underlying molecular mechanism remained elusive. In this study, we demonstrate that Ca(2+) binding and myristoylation independently affect the conformation and functions of human CHP3. Ca(2+) binding increased local flexibility and hydrophobicity of CHP3 indicative of an open conformation. The Ca(2+)-bound CHP3 exhibited a higher affinity for NHE1 and associated stronger with lipid membranes compared to the Mg(2+)-bound CHP3, which adopted a closed conformation. Myristoylation enhanced the local flexibility of CHP3 and decreased its affinity to NHE1 independently of the bound ion, but did not affect its binding to lipid membranes. The data exclude the proposed Ca(2+)-myristoyl switch for CHP3. Instead, a Ca(2+)-independent exposure of the myristoyl moiety is induced by binding of the target peptide to CHP3 enhancing its association to lipid membranes. We name this novel regulatory mechanism 'target-myristoyl switch'. Collectively, the interplay of Ca(2+) binding, myristoylation, and target binding allows for a context-specific regulation of CHP3 functions.
Analysis of Yeast Peroxisomes via Spatial Proteomics. Das, Hirak; Zografakis, Alexandros; Oeljeklaus, Silke; Warscheid, Bettina (2023). 2643 13–31.
Peroxisomes are ubiquitous organelles with essential functions in numerous cellular processes such as lipid metabolism, detoxification of reactive oxygen species, and signaling. Knowledge of the peroxisomal proteome including multi-localized proteins and, most importantly, changes of its composition induced by altering cellular conditions or impaired peroxisome biogenesis and function is of paramount importance for a holistic view on peroxisomes and their diverse functions in a cellular context. In this chapter, we provide a spatial proteomics protocol specifically tailored to the analysis of the peroxisomal proteome of baker's yeast that enables the definition of the peroxisomal proteome under distinct conditions and to monitor dynamic changes of the proteome including the relocation of individual proteins to a different cellular compartment. The protocol comprises subcellular fractionation by differential centrifugation followed by Nycodenz density gradient centrifugation of a crude peroxisomal fraction, quantitative mass spectrometric measurements of subcellular and density gradient fractions, and advanced computational data analysis, resulting in the establishment of organellar maps on a global scale.
COX17 Acetylation via MOF-KANSL Complex Promotes Mitochondrial Integrity and Function. Guhathakurta, Sukanya; Erdogdu, Niyazi Umut; Hoffmann, Juliane J.; Grzadzielewska, Iga; Schendzielorz, Alexander; Seyfferth, Janine; Maartensson, Christoph U.; Corrado, Mauro; Karoutas, Adam; Warscheid, Bettina; Pfanner, Nikolaus; Becker, Thomas; Akhtar, Asifa (2023). 5(11) 1931–1952.
Reversible acetylation of mitochondrial proteins is a regulatory mechanism central to adaptive metabolic responses. Yet, how such functionally relevant protein acetylation is achieved remains unexplored. Here we reveal an unprecedented role of the MYST family lysine acetyltransferase MOF in energy metabolism via mitochondrial protein acetylation. Loss of MOF-KANSL complex members leads to mitochondrial defects including fragmentation, reduced cristae density and impaired mitochondrial electron transport chain complex IV integrity in primary mouse embryonic fibroblasts. We demonstrate COX17, a complex IV assembly factor, as a bona fide acetylation target of MOF. Loss of COX17 or expression of its non-acetylatable mutant phenocopies the mitochondrial defects observed upon MOF depletion. The acetylation-mimetic COX17 rescues these defects and maintains complex IV activity even in the absence of MOF, suggesting an activatory role of mitochondrial electron transport chain protein acetylation. Fibroblasts from patients with MOF syndrome who have intellectual disability also revealed respiratory defects that could be restored by alternative oxidase, acetylation-mimetic COX17 or mitochondrially targeted MOF. Overall, our findings highlight the critical role of MOF-KANSL complex in mitochondrial physiology and provide new insights into MOF syndrome.
ADGym: Design Choices for Deep Anomaly Detection. Jiang, Minqi; Hou, Chaochuan; Zheng, Ao; Han, Songqiao; Huang, Hailiang; Wen, Qingsong; Hu, Xiyang; Zhao, Yue (2023).
Immunoproteasome-Specific Subunit PSMB9 Induction Is Required to Regulate Cellular Proteostasis upon Mitochondrial Dysfunction. Kim, Minji; Serwa, Remigiusz A.; Samluk, Lukasz; Suppanz, Ida; Kodroń, Agata; Stk epkowski, Tomasz M.; Elancheliyan, Praveenraj; Tsegaye, Biniyam; Oeljeklaus, Silke; Wasilewski, Michal; Warscheid, Bettina; Chacinska, Agnieszka (2023). 14(1) 4092.
Perturbed cellular protein homeostasis (proteostasis) and mitochondrial dysfunction play an important role in neurodegenerative diseases, however, the interplay between these two phenomena remains unclear. Mitochondrial dysfunction leads to a delay in mitochondrial protein import, causing accumulation of non-imported mitochondrial proteins in the cytosol and challenging proteostasis. Cells respond by increasing proteasome activity and molecular chaperones in yeast and C. elegans. Here, we demonstrate that in human cells mitochondrial dysfunction leads to the upregulation of a chaperone HSPB1 and, interestingly, an immunoproteasome-specific subunit PSMB9. Moreover, PSMB9 expression is dependent on the translation elongation factor EEF1A2. These mechanisms constitute a defense response to preserve cellular proteostasis under mitochondrial stress. Our findings define a mode of proteasomal activation through the change in proteasome composition driven by EEF1A2 and its spatial regulation, and are useful to formulate therapies to prevent neurodegenerative diseases.
Phosphorylation of the Receptor Protein Pex5p Modulates Import of Proteins into Peroxisomes. Fischer, Sven; Bürgi, Jérôme; Gabay-Maskit, Shiran; Maier, Renate; Mastalski, Thomas; Yifrach, Eden; Obarska-Kosinska, Agnieszka; Rudowitz, Markus; Erdmann, Ralf; Platta, Harald W.; Wilmanns, Matthias; Schuldiner, Maya; Zalckvar, Einat; Oeljeklaus, Silke; Drepper, Friedel; Warscheid, Bettina (2023). 404(2-3) 135–155.
Peroxisomes are organelles with vital functions in metabolism and their dysfunction is associated with human diseases. To fulfill their multiple roles, peroxisomes import nuclear-encoded matrix proteins, most carrying a peroxisomal targeting signal (PTS) 1. The receptor Pex5p recruits PTS1-proteins for import into peroxisomes; whether and how this process is posttranslationally regulated is unknown. Here, we identify 22 phosphorylation sites of Pex5p. Yeast cells expressing phospho-mimicking Pex5p-S507/523D (Pex5p(2D)) show decreased import of GFP with a PTS1. We show that the binding affinity between a PTS1-protein and Pex5p(2D) is reduced. An in vivo analysis of the effect of the phospho-mimicking mutant on PTS1-proteins revealed that import of most, but not all, cargos is affected. The physiological effect of the phosphomimetic mutations correlates with the binding affinity of the corresponding extended PTS1-sequences. Thus, we report a novel Pex5p phosphorylation-dependent mechanism for regulating PTS1-protein import into peroxisomes. In a broader view, this suggests that posttranslational modifications can function in fine-tuning the peroxisomal protein composition and, thus, cellular metabolism.
A Newly Identified Secreted Larval Antigen Elicits Basophil-Dependent Protective Immunity against N. Brasiliensis Infection. Thuma, Natalie; Döhler, Daniela; Mielenz, Dirk; Sticht, Heinrich; Radtke, Daniel; Reimann, Lena; Warscheid, Bettina; Voehringer, David (2022). 13 979491.
Hookworms infect more that 400 million people and cause significant socio-economic burden on endemic countries. The lack of efficient vaccines and the emergence of anthelminthic drug resistance are of major concern. Free-living hookworm larvae infect their hosts via the skin and live as adult worms in the small intestine where they feed on host tissue and blood. Excretory/secretory (E/S) products, released by helminths as they migrate through their host, are thought to play a key role in facilitating infection and successful establishment of parasitism. However, E/S products can also elicit protective immune responses that might be harnessed for vaccine development. By performing Western blots with serum of Nippostrongylus brasiliensis (Nb) infected mice as a model for human hookworm infection, we identified a largely overlapping set of IgG1- and IgE-reactive antigens in E/S from infective L3 stage larvae. Mass spectrometry analysis led to the identification of a new protein family with 6 paralogues in the Nb genome which we termed Nb-LSA1 for "Nippostrongylus brasiliensis larval secreted protein 1". The recombinantly expressed 17 kDa family member Nb-LSA1a was recognized by antibodies in the serum of Nb immune mice. Immunization of mice with Nb-LSA1a in alum elicited a strong IgG1 response but no detectable antigen-specific IgE. Most importantly, immunized mice were largely protected against a challenge Nb infection. This effect was dependent on the presence of basophils and occurred before the parasites reached the intestine. Therefore, basophils appear to play a critical role for rapid control of infection with L3 stage larvae in mice immunized with a single secreted larval protein. A better understanding of basophil-mediated protective immunity and identification of potent larval antigens of human hookworms could help to develop promising vaccination strategies.
The Endoplasmic Reticulum Membrane Protein Complex Localizes to the Mitochondrial - Endoplasmic Reticulum Interface and Its Subunits Modulate Phospholipid Biosynthesis in Trypanosoma Brucei. Iyer, Advaitha; Niemann, Moritz; Serricchio, Mauro; Dewar, Caroline E.; Oeljeklaus, Silke; Farine, Luce; Warscheid, Bettina; Schneider, André; Bütikofer, Peter (2022). 18(5) e1009717.
The endoplasmic reticulum membrane complex (EMC) is a versatile complex that plays a key role in membrane protein biogenesis in the ER. Deletion of the complex has wide-ranging consequences including ER stress, disturbance in lipid transport and organelle tethering, among others. Here we report the function and organization of the evolutionarily conserved EMC (TbEMC) in the highly diverged eukaryote, Trypanosoma brucei. Using (co-) immunoprecipitation experiments in combination with mass spectrometry and whole cell proteomic analyses of parasites after depletion of select TbEMC subunits, we demonstrate that the TbEMC is composed of 9 subunits that are present in a high molecular mass complex localizing to the mitochondrial-endoplasmic reticulum interface. Knocking out or knocking down of single TbEMC subunits led to growth defects of T. brucei procyclic forms in culture. Interestingly, we found that depletion of individual TbEMC subunits lead to disruption of de novo synthesis of phosphatidylcholine (PC) or phosphatidylethanolamine (PE), the two most abundant phospholipid classes in T. brucei. Downregulation of TbEMC1 or TbEMC3 inhibited formation of PC while depletion of TbEMC8 inhibited PE synthesis, pointing to a role of the TbEMC in phospholipid synthesis. In addition, we found that in TbEMC7 knock-out parasites, TbEMC3 is released from the complex, implying that TbEMC7 is essential for the formation or the maintenance of the TbEMC.
Characterization of a Highly Diverged Mitochondrial ATP Synthase F(o) Subunit in Trypanosoma Brucei. Dewar, Caroline E.; Oeljeklaus, Silke; Wenger, Christoph; Warscheid, Bettina; Schneider, André (2022). 298(4) 101829.
The mitochondrial F(1)F(o) ATP synthase of the parasite Trypanosoma brucei has been previously studied in detail. This unusual enzyme switches direction in functionality during the life cycle of the parasite, acting as an ATP synthase in the insect stages, and as an ATPase to generate mitochondrial membrane potential in the mammalian bloodstream stages. Whereas the trypanosome F(1) moiety is relatively highly conserved in structure and composition, the F(o) subcomplex and the peripheral stalk have been shown to be more variable. Interestingly, a core subunit of the latter, the normally conserved subunit b, has been resistant to identification by sequence alignment or biochemical methods. Here, we identified a 17~kDa mitochondrial protein of the inner membrane, Tb927.8.3070, that is essential for normal growth, efficient oxidative phosphorylation, and membrane potential maintenance. Pull-down experiments and native PAGE analysis indicated that the protein is both associated with the F(1)F(o) ATP synthase and integral to its assembly. In addition, its knockdown reduced the levels of F(o) subunits, but not those of F(1), and disturbed the cell cycle. Finally, analysis of structural homology using the HHpred algorithm showed that this protein has structural similarities to F(o) subunit b of other species, indicating that this subunit may be a highly diverged form of the elusive subunit b.
Quantitative Proteomics Identifies the Universally Conserved ATPase Ola1p as a Positive Regulator of Heat Shock Response in Saccharomyces Cerevisiae. Dannenmaier, Stefan; Desroches Altamirano, Christine; Schüler, Lisa; Zhang, Ying; Hummel, Johannes; Milanov, Martin; Oeljeklaus, Silke; Koch, Hans-Georg; Rospert, Sabine; Alberti, Simon; Warscheid, Bettina (2021). 297(5) 101050.
The universally conserved P-loop ATPase Ola1 is implicated in various cellular stress response pathways, as well as in cancer and tumor progression. However, Ola1p functions are divergent between species, and the involved mechanisms are only poorly understood. Here, we studied the role of Ola1p in the heat shock response of the yeast Saccharomyces cerevisiae using a combination of quantitative and pulse labeling-based proteomics approaches, in~vitro studies, and cell-based assays. Our data show that when heat stress is applied to cells lacking Ola1p, the expression of stress-protective proteins is enhanced. During heat stress Ola1p associates with detergent-resistant protein aggregates and rapidly forms assemblies that localize to stress granules. The assembly of Ola1p was also observed in~vitro using purified protein and conditions, which resembled those in living cells. We show that loss of Ola1p results in increased protein ubiquitination of detergent-insoluble aggregates recovered from heat-shocked cells. When cells lacking Ola1p were subsequently relieved from heat stress, reinitiation of translation was delayed, whereas, at the same time, de novo synthesis of central factors required for protein refolding and the clearance of aggregates was enhanced when compared with wild-type cells. The combined data suggest that upon acute heat stress, Ola1p is involved in the stabilization of misfolded proteins, which become sequestered in cytoplasmic stress granules. This function of Ola1p enables cells to resume translation in a timely manner as soon as heat stress is relieved.
Order from Disorder in the Sarcomere: FATZ Forms a Fuzzy but Tight Complex and Phase-Separated Condensates with \($\alpha$\)-Actinin. Sponga, Antonio; Arolas, Joan L.; Schwarz, Thomas C.; Jeffries, Cy M.; Rodriguez Chamorro, Ariadna; Kostan, Julius; Ghisleni, Andrea; Drepper, Friedel; Polyansky, Anton; De Almeida Ribeiro, Euripedes; Pedron, Miriam; Zawadzka-Kazimierczuk, Anna; Mlynek, Georg; Peterbauer, Thomas; Doto, Pierantonio; Schreiner, Claudia; Hollerl, Eneda; Mateos, Borja; Geist, Leonhard; Faulkner, Georgine; Kozminski, Wiktor; Svergun, Dmitri I.; Warscheid, Bettina; Zagrovic, Bojan; Gautel, Mathias; Konrat, Robert; Djinovi’c-Carugo, Kristina (2021). 7(22)
In sarcomeres, \($\alpha$\)-actinin cross-links actin filaments and anchors them to the Z-disk. FATZ (filamin-, \($\alpha$\)-actinin-, and telethonin-binding protein of the Z-disk) proteins interact with \($\alpha$\)-actinin and other core Z-disk proteins, contributing to myofibril assembly and maintenance. Here, we report the first structure and its cellular validation of \($\alpha$\)-actinin-2 in complex with a Z-disk partner, FATZ-1, which is best described as a conformational ensemble. We show that FATZ-1 forms a tight fuzzy complex with \($\alpha$\)-actinin-2 and propose an interaction mechanism via main molecular recognition elements and secondary binding sites. The obtained integrative model reveals a polar architecture of the complex which, in combination with FATZ-1 multivalent scaffold function, might organize interaction partners and stabilize \($\alpha$\)-actinin-2 preferential orientation in Z-disk. Last, we uncover FATZ-1 ability to phase-separate and form biomolecular condensates with \($\alpha$\)-actinin-2, raising the question whether FATZ proteins can create an interaction hub for Z-disk proteins through membraneless compartmentalization during myofibrillogenesis.
Author Correction: Mitochondrial Proteins: From Biogenesis to Functional Networks. Pfanner, Nikolaus; Warscheid, Bettina; Wiedemann, Nils (2021). 22(5) 367.
Quantitative Proteomics Identifies PTP1B as Modulator of B Cell Antigen Receptor Signaling. Schwarz, Jennifer J.; Grundmann, Lorenz; Kokot, Thomas; Kläsener, Kathrin; Fotteler, Sandra; Medgyesi, David; Köhn, Maja; Reth, Michael; Warscheid, Bettina (2021). 4(11)
B cell antigen receptor (BCR) signaling is initiated by protein kinases and limited by counteracting phosphatases that currently are less well studied in their regulation of BCR signaling. Here, we used the B cell line Ramos to identify and quantify human B cell signaling components. Specifically, a protein tyrosine phosphatase profiling revealed a high expression of the protein tyrosine phosphatase 1B (PTP1B) in Ramos and human naïve B cells. The loss of PTP1B leads to increased B cell activation. Through substrate trapping in combination with quantitative mass spectrometry, we identified 22 putative substrates or interactors of PTP1B. We validated Ig\($\alpha$\), CD22, PLC\($\gamma$\)1/2, CBL, BCAP, and APLP2 as specific substrates of PTP1B in Ramos B cells. The tyrosine kinase BTK and the two adaptor proteins GRB2 and VAV1 were identified as direct binding partners and potential substrates of PTP1B. We showed that PTP1B dephosphorylates the inhibitory receptor protein CD22 at phosphotyrosine 807. We conclude that PTP1B negatively modulates BCR signaling by dephosphorylating distinct phosphotyrosines in B cell-specific receptor proteins and various downstream signaling components.
Defining the Interactome of the Human Mitochondrial Ribosome Identifies SMIM4 and TMEM223 as Respiratory Chain Assembly Factors. Dennerlein, Sven; Poerschke, Sabine; Oeljeklaus, Silke; Wang, Cong; Richter-Dennerlein, Ricarda; Sattmann, Johannes; Bauermeister, Diana; Hanitsch, Elisa; Stoldt, Stefan; Langer, Thomas; Jakobs, Stefan; Warscheid, Bettina; Rehling, Peter (2021). 10
Human mitochondria express a genome that encodes thirteen core subunits of the oxidative phosphorylation system (OXPHOS). These proteins insert into the inner membrane co-translationally. Therefore, mitochondrial ribosomes engage with the OXA1L-insertase and membrane-associated proteins, which support membrane insertion of translation products and early assembly steps into OXPHOS complexes. To identify ribosome-associated biogenesis factors for the OXPHOS system, we purified ribosomes and associated proteins from mitochondria. We identified TMEM223 as a ribosome-associated protein involved in complex IV biogenesis. TMEM223 stimulates the translation of COX1 mRNA and is a constituent of early COX1 assembly intermediates. Moreover, we show that SMIM4 together with C12ORF73 interacts with newly synthesized cytochrome b to support initial steps of complex III biogenesis in complex with UQCC1 and UQCC2. Our analyses define the interactome of the human mitochondrial ribosome and reveal novel assembly factors for complex III and IV biogenesis that link early assembly stages to the translation machinery.
DNA Repair Protein APE1 Degrades Dysfunctional Abasic mRNA in Mitochondria Affecting Oxidative Phosphorylation. Barchiesi, Arianna; Bazzani, Veronica; Jabczynska, Agata; Borowski, Lukasz S.; Oeljeklaus, Silke; Warscheid, Bettina; Chacinska, Agnieszka; Szczesny, Roman J.; Vascotto, Carlo (2021). 433(18) 167125.
APE1 is a multifunctional protein which plays a central role in the maintenance of nuclear and mitochondrial genomes repairing DNA lesions caused by oxidative and alkylating agents. In addition, it works as a redox signaling protein regulating gene expression by interacting with many transcriptional factors. Apart from these canonical activities, recent studies have shown that APE1 is also enzymatically active on RNA molecules. The present study unveils for the first time a new role of the mitochondrial form of APE1 protein in the metabolism of RNA in mitochondria. Our data demonstrate that APE1 is associated with mitochondrial messenger RNA and exerts endoribonuclease activity on abasic sites. Loss of APE1 results in the accumulation of damaged mitochondrial mRNA species, determining impairment in protein translation and reduced expression of mitochondrial-encoded proteins, finally leading to less efficient mitochondrial respiration. Altogether, our data demonstrate that APE1 plays an active role in the degradation of the mitochondrial mRNA and has a profound impact on mitochondrial well-being.
Quantitative High-Confidence Human Mitochondrial Proteome and Its Dynamics in Cellular Context. Morgenstern, Marcel; Peikert, Christian D.; Lübbert, Philipp; Suppanz, Ida; Klemm, Cinzia; Alka, Oliver; Steiert, Conny; Naumenko, Nataliia; Schendzielorz, Alexander; Melchionda, Laura; Mühlhäuser, Wignand W. D.; Knapp, Bettina; Busch, Jakob D.; Stiller, Sebastian B.; Dannenmaier, Stefan; Lindau, Caroline; Licheva, Mariya; Eickhorst, Christopher; Galbusera, Riccardo; Zerbes, Ralf M.; Ryan, Michael T.; Kraft, Claudine; Kozjak-Pavlovic, Vera; Drepper, Friedel; Dennerlein, Sven; Oeljeklaus, Silke; Pfanner, Nikolaus; Wiedemann, Nils; Warscheid, Bettina (2021). 33(12) 2464–2483.e18.
Mitochondria are key organelles for cellular energetics, metabolism, signaling, and quality control and have been linked to various diseases. Different views exist on the composition of the human mitochondrial proteome. We classified \($>$\)8,000 proteins in mitochondrial preparations of human cells and defined a mitochondrial high-confidence proteome of \($>$\)1,100 proteins (MitoCoP). We identified interactors of translocases, respiratory chain, and ATP synthase assembly factors. The abundance of MitoCoP proteins covers six orders of magnitude and amounts to 7% of the cellular proteome with the chaperones HSP60-HSP10 being the most abundant mitochondrial proteins. MitoCoP dynamics spans three orders of magnitudes, with half-lives from hours to months, and suggests a rapid regulation of biosynthesis and assembly processes. 460 MitoCoP genes are linked to human diseases with a strong prevalence for the central nervous system and metabolism. MitoCoP will provide a high-confidence resource for placing dynamics, functions, and dysfunctions of mitochondria into the cellular context.
DIMA: Data-Driven Selection of an Imputation Algorithm. Egert, Janine; Brombacher, Eva; Warscheid, Bettina; Kreutz, Clemens (2021). 20(7) 3489–3496.
Imputation is a prominent strategy when dealing with missing values (MVs) in proteomics data analysis pipelines. However, it is difficult to assess the performance of different imputation methods and varies strongly depending on data characteristics. To overcome this issue, we present the concept of a data-driven selection of an imputation algorithm (DIMA). The performance and broad applicability of DIMA are demonstrated on 142 quantitative proteomics data sets from the PRoteomics IDEntifications (PRIDE) database and on simulated data consisting of 5-50% MVs with different proportions of missing not at random and missing completely at random values. DIMA reliably suggests a high-performing imputation algorithm, which is always among the three best algorithms and results in a root mean square error difference (\($\Delta$\)RMSE) \($\leq$\) 10% in 80% of the cases. DIMA implementation is available in MATLAB at github.com/kreutz-lab/OmicsData and in R at github.com/kreutz-lab/DIMAR.
2nSILAC for Quantitative Proteomics of Prototrophic Baker’s Yeast. Dannenmaier, Stefan; Oeljeklaus, Silke; Warscheid, Bettina (2021). (Vol. 2228) 253–270.
Stable isotope labeling by amino acids in cell culture (SILAC) combined with high-resolution mass spectrometry is a quantitative strategy for the comparative analysis of (sub)proteomes. It is based on the metabolic incorporation of stable isotope-coded amino acids during growth of cells or organisms. Here, complete labeling of proteins with the amino acid(s) selected for incorporation needs to be guaranteed to enable accurate quantification on a proteomic scale. Wild-type strains of baker's yeast (Saccharomyces cerevisiae ), which is a widely accepted and well-studied eukaryotic model organism, are generally able to synthesize all amino acids on their own (i.e., prototrophic). To render them amenable to SILAC, auxotrophies are introduced by genetic manipulations. We addressed this limitation by developing a generic strategy for complete "native" labeling of prototrophic S. cerevisiae with isotope-coded arginine and lysine, referred to as "2nSILAC". It allows for directly using and screening several genome-wide yeast mutant collections that are easily accessible to the scientific community for functional proteomic studies but are based on prototrophic variants of S. cerevisiae.
Molecular Basis of F-actin Regulation and Sarcomere Assembly via Myotilin. Kostan, Julius; Pavv siv c, Miha; Puv z, Vid; Schwarz, Thomas C.; Drepper, Friedel; Molt, Sibylle; Graewert, Melissa Ann; Schreiner, Claudia; Sajko, Sara; van der Ven, Peter F. M.; Onipe, Adekunle; Svergun, Dmitri I.; Warscheid, Bettina; Konrat, Robert; Fürst, Dieter O.; Lenarv civ c, Brigita; Djinovi’c-Carugo, Kristina (2021). 19(4) e3001148.
Sarcomeres, the basic contractile units of striated muscle cells, contain arrays of thin (actin) and thick (myosin) filaments that slide past each other during contraction. The Ig-like domain-containing protein myotilin provides structural integrity to Z-discs-the boundaries between adjacent sarcomeres. Myotilin binds to Z-disc components, including F-actin and \($\alpha$\)-actinin-2, but the molecular mechanism of binding and implications of these interactions on Z-disc integrity are still elusive. To illuminate them, we used a combination of small-angle X-ray scattering, cross-linking mass spectrometry, and biochemical and molecular biophysics approaches. We discovered that myotilin displays conformational ensembles in solution. We generated a structural model of the F-actin:myotilin complex that revealed how myotilin interacts with and stabilizes F-actin via its Ig-like domains and flanking regions. Mutant myotilin designed with impaired F-actin binding showed increased dynamics in cells. Structural analyses and competition assays uncovered that myotilin displaces tropomyosin from F-actin. Our findings suggest a novel role of myotilin as a co-organizer of Z-disc assembly and advance our mechanistic understanding of myotilin's structural role in Z-discs.
COA6 Facilitates Cytochrome c Oxidase Biogenesis as Thiol-reductase for Copper Metallochaperones in Mitochondria. Pacheu-Grau, David; Wasilewski, Michal; Oeljeklaus, Silke; Gibhardt, Christine Silvia; Aich, Abhishek; Chudenkova, Margarita; Dennerlein, Sven; Deckers, Markus; Bogeski, Ivan; Warscheid, Bettina; Chacinska, Agnieszka; Rehling, Peter (2020). 432(7) 2067–2079.
The mitochondrial cytochrome c oxidase, the terminal enzyme of the respiratory chain, contains heme and copper centers for electron transfer. The conserved COX2 subunit contains the Cu(A) site, a binuclear copper center. The copper chaperones SCO1, SCO2, and COA6, are required for Cu(A) center formation. Loss of function of these chaperones and the concomitant cytochrome c oxidase deficiency cause severe human disorders. Here we analyzed the molecular function of COA6 and the consequences of COA6 deficiency for mitochondria. Our analyses show that loss of COA6 causes combined complex I and complex IV deficiency and impacts membrane potential-driven protein transport across the inner membrane. We demonstrate that COA6 acts as a thiol-reductase to reduce disulfide bridges of critical cysteine residues in SCO1 and SCO2. Cysteines within the CX(3)CX(N)H domain of SCO2 mediate its interaction with COA6 but are dispensable for SCO2-SCO1 interaction. Our analyses define COA6 as thiol-reductase, which is essential for Cu(A) biogenesis.
Mitochondrial Proteins: From Biogenesis to Functional Networks. Pfanner, Nikolaus; Warscheid, Bettina; Wiedemann, Nils (2019). 20(5) 267–284.
Mitochondria are essential for the viability of eukaryotic cells as they perform crucial functions in bioenergetics, metabolism and signalling and have been associated with numerous diseases. Recent functional and proteomic studies have revealed the remarkable complexity of mitochondrial protein organization. Protein machineries with diverse functions such as protein translocation, respiration, metabolite transport, protein quality control and the control of membrane architecture interact with each other in dynamic networks. In this Review, we discuss the emerging role of the mitochondrial protein import machinery as a key organizer of these mitochondrial protein networks. The preprotein translocases that reside on the mitochondrial membranes not only function during organelle biogenesis to deliver newly synthesized proteins to their final mitochondrial destination but also cooperate with numerous other mitochondrial protein complexes that perform a wide range of functions. Moreover, these protein networks form membrane contact sites, for example, with the endoplasmic reticulum, that are key for integration of mitochondria with cellular function, and defects in protein import can lead to diseases.
Noncompetitive Binding of PpiD and YidC to the SecYEG Translocon Expands the Global View on the SecYEG Interactome in Escherichia Coli. Jauss, Benjamin; Petriman, Narcis-Adrian; Drepper, Friedel; Franz, Lisa; Sachelaru, Ilie; Welte, Thomas; Steinberg, Ruth; Warscheid, Bettina; Koch, Hans-Georg (2019). 294(50) 19167–19183.
The SecYEG translocon constitutes the major protein transport channel in bacteria and transfers an enormous variety of different secretory and inner-membrane proteins. The minimal core of the SecYEG translocon consists of three inner-membrane proteins, SecY, SecE, and SecG, which, together with appropriate targeting factors, are sufficient for protein transport in vitro However, in vivo the SecYEG translocon has been shown to associate with multiple partner proteins, likely allowing the SecYEG translocon to process its diverse substrates. To obtain a global view on SecYEG plasticity in Escherichia coli, here we performed a quantitative interaction proteomic analysis, which identified several known SecYEG-interacting proteins, verified the interaction of SecYEG with quality-control proteins, and revealed several previously unknown putative SecYEG-interacting proteins. Surprisingly, we found that the chaperone complex PpiD/YfgM is the most prominent interaction partner of SecYEG. Detailed analyses of the PpiD-SecY interaction by site-directed cross-linking revealed that PpiD and the established SecY partner protein YidC use almost completely-overlapping binding sites on SecY. Both PpiD and YidC contacted the lateral gate, the plug domain, and the periplasmic cavity of SecY. However, quantitative MS and cross-linking analyses revealed that despite having almost identical binding sites, their binding to SecY is noncompetitive. This observation suggests that the SecYEG translocon forms different substrate-independent subassemblies in which SecYEG either associates with YidC or with the PpiD/YfgM complex. In summary, the results of this study indicate that the PpiD/YfgM chaperone complex is a primary interaction partner of the SecYEG translocon.
Mitochondrial Protein Translocation-Associated Degradation. Maartensson, Christoph U.; Priesnitz, Chantal; Song, Jiyao; Ellenrieder, Lars; Doan, Kim Nguyen; Boos, Felix; Floerchinger, Alessia; Zufall, Nicole; Oeljeklaus, Silke; Warscheid, Bettina; Becker, Thomas (2019). 569(7758) 679–683.
Mitochondrial biogenesis and functions depend on the import of precursor proteins via the 'translocase of the outer membrane' (TOM complex). Defects in protein import lead to an accumulation of mitochondrial precursor proteins that induces a range of cellular stress responses. However, constitutive quality-control mechanisms that clear trapped precursor proteins from the TOM channel under non-stress conditions have remained unknown. Here we report that in Saccharomyces cerevisiae Ubx2, which functions in endoplasmic reticulum-associated degradation, is crucial for this quality-control process. A pool of Ubx2 binds to the TOM complex to recruit the AAA ATPase Cdc48 for removal of arrested precursor proteins from the TOM channel. This mitochondrial protein translocation-associated degradation (mitoTAD) pathway continuously monitors the TOM complex under non-stress conditions to prevent clogging of the TOM channel with precursor proteins. The mitoTAD pathway ensures that mitochondria maintain their full protein-import capacity, and protects cells against proteotoxic stress induced by impaired transport of proteins into mitochondria.
The Highly Diverged Trypanosomal MICOS Complex Is Organized in a Nonessential Integral Membrane and an Essential Peripheral Module. Eichenberger, Claudia; Oeljeklaus, Silke; Bruggisser, Julia; Mani, Jan; Haenni, Beat; Kaurov, Iosif; Niemann, Moritz; Zuber, Benoît; Lukev s, Julius; Hashimi, Hassan; Warscheid, Bettina; Schimanski, Bernd; Schneider, André (2019). 112(6) 1731–1743.
The mitochondrial contact site and cristae organization system (MICOS) mediates the formation of cristae, invaginations in the mitochondrial inner membrane. The highly diverged MICOS complex of the parasitic protist Trypanosoma brucei consists of nine subunits. Except for two Mic10-like and a Mic60-like protein, all subunits are specific for kinetoplastids. Here, we determined on a proteome-wide scale how ablation of individual MICOS subunits affects the levels of the other subunits. The results reveal co-regulation of TbMic10-1, TbMic10-2, TbMic16 and TbMic60, suggesting that these nonessential, integral inner membrane proteins form an interdependent network. Moreover, the ablation of TbMic34 and TbMic32 reveals another network consisting of the essential, intermembrane space-localized TbMic20, TbMic32, TbMic34 and TbMic40, all of which are peripherally associated with the inner membrane. The downregulation of TbMic20, TbMic32 and TbMic34 also interferes with mitochondrial protein import and reduces the size of the TbMic10-containing complexes. Thus, the diverged MICOS of trypanosomes contains two subcomplexes: a nonessential membrane-integrated one, organized around the conserved Mic10 and Mic60, that mediates cristae formation, and an essential membrane-peripheral one consisting of four kinetoplastid-specific subunits, that is required for import of intermembrane space proteins.
The Diverged Trypanosome MICOS Complex as a Hub for Mitochondrial Cristae Shaping and Protein Import. Kaurov, Iosif; Vancová, Marie; Schimanski, Bernd; Cadena, Lawrence Rudy; Heller, Jiv rí; Bíl’y, Tomáv s; Potv ev sil, David; Eichenberger, Claudia; Bruce, Hannah; Oeljeklaus, Silke; Warscheid, Bettina; Zdráhal, Zbynv ek; Schneider, André; Lukev s, Julius; Hashimi, Hassan (2018). 28(21) 3393–3407.e5.
The mitochondrial contact site and cristae organization system (MICOS) is a multiprotein complex responsible for cristae formation. Even though cristae are found in all mitochondria capable of oxidative phosphorylation, only Mic10 and Mic60 appear to be conserved throughout eukaryotes. The remaining 4 or 5 known MICOS subunits are specific to the supergroup Opisthokonta, which includes yeast and mammals that are the only organisms in which this complex has been analyzed experimentally. We have isolated the MICOS from Trypanosoma brucei, a member of the supergroup Excavata that is profoundly diverged from opisthokonts. We show that it is required for the maintenance of the unique discoidal cristae that typify excavates, such as euglenids and kinetoplastids, the latter of which include trypanosomes. The trypanosome MICOS consists of 9 subunits, most of which are essential for normal growth. Unlike in opisthokonts, it contains two distinct Mic10 orthologs and an unconventional putative Mic60 that lacks a mitofilin domain. Interestingly, one of the essential trypanosomatid-specific MICOS subunits called TbMic20 is a thioredoxin-like protein that appears to be involved in import of intermembrane space proteins, including respiratory chain complex assembly factors. This result points to trypanosome MICOS coordinating cristae shaping and population of its membrane with proteins involved in respiration, the latter via the catalytic activity of TbMic20. Thus, trypanosome MICOS allows us to define which of its features are conserved in all eukaryotes and decipher those that represent lineage-specific adaptations.
Complete Native Stable Isotope Labeling by Amino Acids of Saccharomyces Cerevisiae for Global Proteomic Analysis. Dannenmaier, Stefan; Stiller, Sebastian B.; Morgenstern, Marcel; Lübbert, Philipp; Oeljeklaus, Silke; Wiedemann, Nils; Warscheid, Bettina (2018). 90(17) 10501–10509.
Knowledge about the functions of individual proteins on a system-wide level is crucial to fully understand molecular mechanisms underlying cellular processes. A considerable part of the proteome across all organisms is still poorly characterized. Mass spectrometry is an efficient technology for the global study of proteins. One of the most prominent methods for accurate proteome-wide comparative quantification is stable isotope labeling by amino acids in cell culture (SILAC). However, application of SILAC to prototrophic organisms such as Saccharomyces cerevisiae, also known as baker's yeast, is compromised since they are able to synthesize all amino acids on their own. Here, we describe an advanced strategy, termed 2nSILAC, that allows for in vivo labeling of prototrophic baker's yeast using heavy arginine and lysine under fermentable and respiratory growth conditions, making it a suitable tool for the global study of protein functions. This generic 2nSILAC strategy allows for directly using and systematically screening yeast mutant strain collections available to the scientific community. We exemplarily demonstrate its high potential by analyzing the effects of mitochondrial gene deletions in mitochondrial fractions using quantitative mass spectrometry revealing the role of Coi1 for the assembly of cytochrome c oxidase (respiratory chain complex IV).
Vps39 Interacts with Tom40 to Establish One of Two Functionally Distinct Vacuole-Mitochondria Contact Sites. González Montoro, Ayelén; Auffarth, Kathrin; Hönscher, Carina; Bohnert, Maria; Becker, Thomas; Warscheid, Bettina; Reggiori, Fulvio; van der Laan, Martin; Fröhlich, Florian; Ungermann, Christian (2018). 45(5) 621–636.e7.
The extensive subcellular network of membrane contact sites plays central roles in organelle biogenesis and communication, yet the precise contributions of the involved machineries remain largely enigmatic. The yeast vacuole forms a membrane contact site with mitochondria, called vacuolar and mitochondrial patch (vCLAMP). Formation of vCLAMPs involves the vacuolar Rab GTPase Ypt7 and the Ypt7-interacting Vps39 subunit of the HOPS tethering complex. Here, we uncover the general preprotein translocase of the outer membrane (TOM) subunit Tom40 as the direct binding partner of Vps39 on mitochondria. We identify Vps39 mutants defective in TOM binding, but functional for HOPS. Cells that cannot form vCLAMPs show reduced growth under stress conditions and impaired survival upon starvation. Unexpectedly, our mutant analysis revealed the existence of two functionally independent vacuole-mitochondria MCSs: one formed by the Ypt7-Vps39-Tom40 tether and a second one by Vps13-Mcp1, which is redundant with ER-mitochondrial contacts formed by ERMES.
The Mitochondrial TMEM177 Associates with COX20 during COX2 Biogenesis. Lorenzi, Isotta; Oeljeklaus, Silke; Aich, Abhishek; Ronsör, Christin; Callegari, Sylvie; Dudek, Jan; Warscheid, Bettina; Dennerlein, Sven; Rehling, Peter (2018). 1865(2) 323–333.
The three mitochondrial-encoded proteins, COX1, COX2, and COX3, form the core of the cytochrome c oxidase. Upon synthesis, COX2 engages with COX20 in the inner mitochondrial membrane, a scaffold protein that recruits metallochaperones for copper delivery to the Cu(A)-Site of COX2. Here we identified the human protein, TMEM177 as a constituent of the COX20 interaction network. Loss or increase in the amount of TMEM177 affects COX20 abundance leading to reduced or increased COX20 levels respectively. TMEM177 associates with newly synthesized COX2 and SCO2 in a COX20-dependent manner. Our data shows that by unbalancing the amount of TMEM177, newly synthesized COX2 accumulates in a COX20-associated state. We conclude that TMEM177 promotes assembly of COX2 at the level of Cu(A)-site formation.
Quantitative Proteomics Identifies Redox Switches for Global Translation Modulation by Mitochondrially Produced Reactive Oxygen Species. Topf, Ulrike; Suppanz, Ida; Samluk, Lukasz; Wrobel, Lidia; Böser, Alexander; Sakowska, Paulina; Knapp, Bettina; Pietrzyk, Martyna K.; Chacinska, Agnieszka; Warscheid, Bettina (2018). 9(1) 324.
The generation of reactive oxygen species (ROS) is inevitably linked to life. However, the precise role of ROS in signalling and specific targets is largely unknown. We perform a global proteomic analysis to delineate the yeast redoxome to a depth of more than 4,300 unique cysteine residues in over 2,200 proteins. Mapping of redox-active thiols in proteins exposed to exogenous or endogenous mitochondria-derived oxidative stress reveals ROS-sensitive sites in several components of the translation apparatus. Mitochondria are the major source of cellular ROS. We demonstrate that increased levels of intracellular ROS caused by dysfunctional mitochondria serve as a signal to attenuate global protein synthesis. Hence, we propose a universal mechanism that controls protein synthesis by inducing reversible changes in the translation machinery upon modulating the redox status of proteins involved in translation. This crosstalk between mitochondria and protein synthesis may have an important contribution to pathologies caused by dysfunctional mitochondria.
The Interaction Network of the YidC Insertase with the SecYEG Translocon, SRP and the SRP Receptor FtsY. Petriman, Narcis-Adrian; Jauß, Benjamin; Hufnagel, Antonia; Franz, Lisa; Sachelaru, Ilie; Drepper, Friedel; Warscheid, Bettina; Koch, Hans-Georg (2018). 8(1) 578.
YidC/Oxa1/Alb3 are essential proteins that operate independently or cooperatively with the Sec machinery during membrane protein insertion in bacteria, archaea and eukaryotic organelles. Although the interaction between the bacterial SecYEG translocon and YidC has been observed in multiple studies, it is still unknown which domains of YidC are in contact with the SecYEG translocon. By in vivo and in vitro site-directed and para-formaldehyde cross-linking we identified the auxiliary transmembrane domain 1 of E. coli YidC as a major contact site for SecY and SecG. Additional SecY contacts were observed for the tightly packed globular domain and the C1 loop of YidC, which reveals that the hydrophilic cavity of YidC faces the lateral gate of SecY. Surprisingly, YidC-SecYEG contacts were only observed when YidC and SecYEG were present at about stoichiometric concentrations, suggesting that the YidC-SecYEG contact in vivo is either very transient or only observed for a very small SecYEG sub-population. This is different for the YidC-SRP and YidC-FtsY interaction, which involves the C1 loop of YidC and is efficiently observed even at sub-stoichiometric concentrations of SRP/FtsY. In summary, our data provide a first detailed view on how YidC interacts with the SecYEG translocon and the SRP-targeting machinery.
Saccharomyces Cerevisiae Cells Lacking Pex3 Contain Membrane Vesicles That Harbor a Subset of Peroxisomal Membrane Proteins. Wróblewska, Justyna P.; Cruz-Zaragoza, Luis Daniel; Yuan, Wei; Schummer, Andreas; Chuartzman, Silvia G.; de Boer, Rinse; Oeljeklaus, Silke; Schuldiner, Maya; Zalckvar, Einat; Warscheid, Bettina; Erdmann, Ralf; van der Klei, Ida J. (2017). 1864(10) 1656–1667.
Pex3 has been proposed to be important for the exit of peroxisomal membrane proteins (PMPs) from the ER, based on the observation that PMPs accumulate at the ER in Saccharomyces cerevisiae pex3 mutant cells. Using a combination of microscopy and biochemical approaches, we show that a subset of the PMPs, including the receptor docking protein Pex14, localizes to membrane vesicles in S. cerevisiae pex3 cells. These vesicles are morphologically distinct from the ER and do not co-sediment with ER markers in cell fractionation experiments. At the vesicles, Pex14 assembles with other peroxins (Pex13, Pex17, and Pex5) to form a complex with a composition similar to the PTS1 import pore in wild-type cells. Fluorescence microscopy studies revealed that also the PTS2 receptor Pex7, the importomer organizing peroxin Pex8, the ubiquitin conjugating enzyme Pex4 with its recruiting PMP Pex22, as well as Pex15 and Pex25 co-localize with Pex14. Other peroxins (including the RING finger complex and Pex27) did not accumulate at these structures, of which Pex11 localized to mitochondria. In line with these observations, proteomic analysis showed that in addition to the docking proteins and Pex5, also Pex7, Pex4/Pex22 and Pex25 were present in Pex14 complexes isolated from pex3 cells. However, formation of the entire importomer was not observed, most likely because Pex8 and the RING proteins were absent in the Pex14 protein complexes. Our data suggest that peroxisomal membrane vesicles can form in the absence of Pex3 and that several PMPs can insert in these vesicles in a Pex3 independent manner.
Membrane Localization of Acetylated CNK1 Mediates a Positive Feedback on RAF/ERK Signaling. Fischer, Adrian; Mühlhäuser, Wignand W. D.; Warscheid, Bettina; Radziwill, Gerald (2017). 3(8) e1700475.
Spatiotemporal control is a common mechanism that modulates activity and function of signal transducers in the signaling network. We identified acetylation of CNK1 (connector enhancer of kinase suppressor of Ras-1) as a late step in the activation of CNK1 signaling, accompanied with prolonged stimulation of extracellular signal-regulated kinase (ERK). We identified the acetyltransferase CREB (cyclic adenosine 3',5'-monophosphate response element-binding protein)-binding protein and the deacetylase SIRT2 (sirtuin type 2) as novel binding partners of CNK1, modulating the acetylation state of CNK1. Acetylation of CNK1 at position Lys(414) located in the pleckstrin homology domain drives membrane localization of CNK1 in growth factor-stimulated cells. Inhibition of ERK signaling abolishes CNK1 acetylation. Cosmic database search identified CNK1 mutants at position Arg(426) near the acetylation site in several human tumor types. These mutants show constitutive acetylation and membrane localization. CNK1 mutants substituting Arg(426), the acetylation mimetic mutant CNK1-K414Q, and membrane-anchored CNK1 mutants all interact with the protein kinase CRAF and stimulate ERK-dependent cell proliferation and cell migration. In RAS-transformed cells, CNK1 is acetylated and membrane-bound and drives cell proliferation. Thus, growth factor-stimulated ERK signaling induces CNK1 acetylation, and acetylated CNK1 promotes ERK signaling, demonstrating a novel function of CNK1 as positive feedback regulator of the RAF/MEK (mitogen-activated protein kinase kinase)/ERK pathway. In addition, acetylation of CNK1 is an important step in oncogenic signaling, promoting cell proliferation and migration.
Biogenesis of the Mitochondrial DNA Inheritance Machinery in the Mitochondrial Outer Membrane of Trypanosoma Brucei. Käser, Sandro; Willemin, Mathilde; Schnarwiler, Felix; Schimanski, Bernd; Poveda-Huertes, Daniel; Oeljeklaus, Silke; Haenni, Beat; Zuber, Benoît; Warscheid, Bettina; Meisinger, Chris; Schneider, André (2017). 13(12) e1006808.
Mitochondria cannot form de novo but require mechanisms that mediate their inheritance to daughter cells. The parasitic protozoan Trypanosoma brucei has a single mitochondrion with a single-unit genome that is physically connected across the two mitochondrial membranes with the basal body of the flagellum. This connection, termed the tripartite attachment complex (TAC), is essential for the segregation of the replicated mitochondrial genomes prior to cytokinesis. Here we identify a protein complex consisting of three integral mitochondrial outer membrane proteins-TAC60, TAC42 and TAC40-which are essential subunits of the TAC. TAC60 contains separable mitochondrial import and TAC-sorting signals and its biogenesis depends on the main outer membrane protein translocase. TAC40 is a member of the mitochondrial porin family, whereas TAC42 represents a novel class of mitochondrial outer membrane \($\beta$\)-barrel proteins. Consequently TAC40 and TAC42 contain C-terminal \($\beta$\)-signals. Thus in trypanosomes the highly conserved \($\beta$\)-barrel protein assembly machinery plays a major role in the biogenesis of its unique mitochondrial genome segregation system.
Identification of Novel STAT6-Regulated Proteins in Mouse B Cells by Comparative Transcriptome and Proteome Analysis. Mokada-Gopal, Lavanya; Boeser, Alexander; Lehmann, Christian H. K.; Drepper, Friedel; Dudziak, Diana; Warscheid, Bettina; Voehringer, David (2017). 198(9) 3737–3745.
The transcription factor STAT6 plays a key role in mediating signaling downstream of the receptors for IL-4 and IL-13. In B cells, STAT6 is required for class switch recombination to IgE and for germinal center formation during type 2 immune responses directed against allergens or helminths. In this study, we compared the transcriptomes and proteomes of primary mouse B cells from wild-type and STAT6-deficient mice cultured for 4 d in the presence or absence of IL-4. Microarray analysis revealed that 214 mRNAs were upregulated and 149 were downregulated \($>$\)3-fold by IL-4 in a STAT6-dependent manner. Across all samples, \($\sim$\)5000 proteins were identified by label-free quantitative liquid chromatography/mass spectrometry. A total of 149 proteins was found to be differentially expressed \($>$\)3-fold between IL-4-stimulated wild-type and STAT6(-/-) B cells (75 upregulated and 74 downregulated). Comparative analysis of the proteome and transcriptome revealed that expression of these proteins was mainly regulated at the transcriptional level, which argues against a major role for posttranscriptional mechanisms that modulate the STAT6-dependent proteome. Nine proteins were selected for confirmation by flow cytometry or Western blot. We show that CD30, CD79b, SLP-76, DEC205, IL-5R\($\alpha$\), STAT5, and Thy1 are induced by IL-4 in a STAT6-dependent manner. In contrast, Syk and Fc receptor-like 1 were downregulated. This dataset provides a framework for further functional analysis of newly identified IL-4-regulated proteins in B cells that may contribute to germinal center formation and IgE switching in type 2 immunity.
INA Complex Liaises the F(1)F(o)-ATP Synthase Membrane Motor Modules. Naumenko, Nataliia; Morgenstern, Marcel; Rucktäschel, Robert; Warscheid, Bettina; Rehling, Peter (2017). 8(1) 1237.
The F(1)F(0)-ATP synthase translates a proton flux across the inner mitochondrial membrane into a mechanical rotation, driving anhydride bond formation in the catalytic portion. The complex's membrane-embedded motor forms a proteinaceous channel at the interface between Atp9 ring and Atp6. To prevent unrestricted proton flow dissipating the H(+)-gradient, channel formation is a critical and tightly controlled step during ATP synthase assembly. Here we show that the INA complex (INAC) acts at this decisive step promoting Atp9-ring association with Atp6. INAC binds to newly synthesized mitochondrial-encoded Atp6 and Atp8 in complex with maturation factors. INAC association is retained until the F(1)-portion is built on Atp6/8 and loss of INAC causes accumulation of the free F(1). An independent complex is formed between INAC and the Atp9 ring. We conclude that INAC maintains assembly intermediates of the F(1) F(0)-ATP synthase in a primed state for the terminal assembly step-motor module formation.
Charting Organellar Importomes by Quantitative Mass Spectrometry. Peikert, Christian D.; Mani, Jan; Morgenstern, Marcel; Käser, Sandro; Knapp, Bettina; Wenger, Christoph; Harsman, Anke; Oeljeklaus, Silke; Schneider, André; Warscheid, Bettina (2017). 8 15272.
Protein import into organelles is essential for all eukaryotes and facilitated by multi-protein translocation machineries. Analysing whether a protein is transported into an organelle is largely restricted to single constituents. This renders knowledge about imported proteins incomplete, limiting our understanding of organellar biogenesis and function. Here we introduce a method that enables charting an organelle's importome. The approach relies on inducible RNAi-mediated knockdown of an essential subunit of a translocase to impair import and quantitative mass spectrometry. To highlight its potential, we established the mitochondrial importome of Trypanosoma brucei, comprising 1,120 proteins including 331 new candidates. Furthermore, the method allows for the identification of proteins with dual or multiple locations and the substrates of distinct protein import pathways. We demonstrate the specificity and versatility of this ImportOmics method by targeting import factors in mitochondria and glycosomes, which demonstrates its potential for globally studying protein import and inventories of organelles.
Expanding the Archaellum Regulatory Network - the Eukaryotic Protein Kinases ArnC and ArnD Influence Motility of Sulfolobus Acidocaldarius. Hoffmann, Lena; Schummer, Andreas; Reimann, Julia; Haurat, Maria F.; Wilson, Amanda J.; Beeby, Morgan; Warscheid, Bettina; Albers, Sonja-V. (2017). 6(1)
Expression of the archaellum, the archaeal-type IV pilus-like rotating motility structure is upregulated under nutrient limitation. This is controlled by a network of regulators, called the archaellum regulatory network (arn). Several of the components of this network in Sulfolobus~acidocaldarius can be phosphorylated, and the deletion of the phosphatase PP2A results in strongly increased motility during starvation, indicating a role for phosphorylation in the regulation of motility. Analysis of the motility of different protein kinase deletion strains revealed that deletion of saci_0965, saci_1181, and saci_1193 resulted in reduced motility, whereas the deletion of saci_1694 resulted in hypermotility. Here ArnC (Saci\_1193) and ArnD (Saci\_1694) are characterized. Purified ArnC and ArnD phosphorylate serine and threonine residues in the C-terminus of the repressor ArnB. arnC is upregulated in starvation medium, whereas arnD is constitutively expressed. However, while differences in the expression and levels of flaB were observed in the \($\Delta$\)arnD strain during growth under rich conditions, under nutrient limiting conditions the \($\Delta$\)arnC and \($\Delta$\)arnD strains showed no large differences in the expression levels of the archaellum or of the studied regulators. This suggests that next to the regulation via the archaellum regulatory network additional regulatory mechanisms of expression and/or activity of the archaellum exist.
Xilmass: A New Approach toward the Identification of Cross-Linked Peptides. Yilmaz, c Sule; Drepper, Friedel; Hulstaert, Niels; v Cerniv c, Mav sa; Gevaert, Kris; Economou, Anastassios; Warscheid, Bettina; Martens, Lennart; Vandermarliere, Elien (2016). 88(20) 9949–9957.
Chemical cross-linking coupled with mass spectrometry plays an important role in unravelling protein interactions, especially weak and transient ones. Moreover, cross-linking complements several structural determination approaches such as cryo-EM. Although several computational approaches are available for the annotation of spectra obtained from cross-linked peptides, there remains room for improvement. Here, we present Xilmass, a novel algorithm to identify cross-linked peptides that introduces two new concepts: (i) the cross-linked peptides are represented in the search database such that the cross-linking sites are explicitly encoded, and (ii) the scoring function derived from the Andromeda algorithm was adapted to score against a theoretical tandem mass spectrometry (MS/MS) spectrum that contains the peaks from all possible fragment ions of a cross-linked peptide pair. The performance of Xilmass was evaluated against the recently published Kojak and the popular pLink algorithms on a calmodulin-plectin complex data set, as well as three additional, published data sets. The results show that Xilmass typically had the highest number of identified distinct cross-linked sites and also the highest number of predicted cross-linked sites.
Identification of Cell Cycle Dependent Interaction Partners of the Septins by Quantitative Mass Spectrometry. Renz, Christian; Oeljeklaus, Silke; Grinhagens, Sören; Warscheid, Bettina; Johnsson, Nils; Gronemeyer, Thomas (2016). 11(2) e0148340.
The septins are a conserved family of GTP-binding proteins that, in the baker's yeast, assemble into a highly ordered array of filaments at the mother bud neck. These filaments undergo significant structural rearrangements during the cell cycle. We aimed at identifying key components that are involved in or regulate the transitions of the septins. By combining cell synchronization and quantitative affinity-purification mass-spectrometry, we performed a screen for specific interaction partners of the septins at three distinct stages of the cell cycle. A total of 83 interaction partners of the septins were assigned. Surprisingly, we detected DNA-interacting/nuclear proteins and proteins involved in ribosome biogenesis and protein synthesis predominantly present in alpha-factor arrested that do not display an assembled septin structure. Furthermore, two distinct sets of regulatory proteins that are specific for cells at S-phase with a stable septin collar or at mitosis with split septin rings were identified. Complementary methods like SPLIFF and immunoprecipitation allowed us to more exactly define the spatial and temporal characteristics of selected hits of the AP-MS screen.
Pex17p-Dependent Assembly of Pex14p/Dyn2p-subcomplexes of the Peroxisomal Protein Import Machinery. Chan, Anna; Schummer, Andreas; Fischer, Sven; Schröter, Thomas; Cruz-Zaragoza, Luis Daniel; Bender, Julian; Drepper, Friedel; Oeljeklaus, Silke; Kunau, Wolf-H.; Girzalsky, Wolfgang; Warscheid, Bettina; Erdmann, Ralf (2016). 95(12) 585–597.
Peroxisomal matrix protein import is facilitated by cycling receptors that recognize their cargo proteins in the cytosol by peroxisomal targeting sequences (PTS). In the following, the assembled receptor-cargo complex is targeted to the peroxisomal membrane where it docks to the docking-complex as part of the peroxisomal translocation machinery. The docking-complex is composed of Pex13p, Pex14p and in yeast also Pex17p, whose function is still elusive. In order to characterize the function of Pex17p, we compared the composition and size of peroxisomal receptor-docking complexes from wild-type and pex17\($\Delta$\) cells. Our data demonstrate that the deficiency of Pex17p affects the stoichiometry of the constituents of an isolated 600kDa complex and that pex17\($\Delta$\) cells lack a high molecular weight complex (\($>$\)900kDa) of unknown function. We identified the dynein light chain protein Dyn2p as an additional core component of the Pex14p/Pex17p-complex. Both, Pex14p and Pex17p interact directly with Dyn2p, but in vivo, Pex17p turned out to be prerequisite for an association of Dyn2p with Pex14p. Finally, like pex17\($\Delta$\) also dyn2\($\Delta$\) cells lack the high molecular weight complex. As dyn2\($\Delta$\) cells also display reduced peroxisomal function, our data indicate that Dyn2p-dependent formation of the high molecular weight Pex14p-complex is required to maintain peroxisomal function on wild-type level.
Mitochondrial OXA Translocase Plays a Major Role in Biogenesis of Inner-Membrane Proteins. Stiller, Sebastian B.; Höpker, Jan; Oeljeklaus, Silke; Schütze, Conny; Schrempp, Sandra G.; Vent-Schmidt, Jens; Horvath, Susanne E.; Frazier, Ann E.; Gebert, Natalia; van der Laan, Martin; Bohnert, Maria; Warscheid, Bettina; Pfanner, Nikolaus; Wiedemann, Nils (2016). 23(5) 901–908.
The mitochondrial inner membrane harbors three protein translocases. Presequence translocase and carrier translocase are essential for importing nuclear-encoded proteins. The oxidase assembly (OXA) translocase is required for exporting mitochondrial-encoded proteins; however, different views exist about its relevance for nuclear-encoded proteins. We report that OXA plays a dual role in the biogenesis of nuclear-encoded mitochondrial proteins. First, a systematic analysis of OXA-deficient mitochondria led to~an unexpected expansion of the spectrum of OXA substrates imported via the presequence pathway. Second, biogenesis of numerous metabolite carriers depends on OXA, although they are not imported by~the presequence pathway. We show that OXA is crucial for the biogenesis of the Tim18-Sdh3 module of the carrier translocase. The export translocase OXA is thus required for the import of metabolite carriers by promoting assembly of the carrier translocase. We conclude that OXA is of central importance for the biogenesis of the mitochondrial inner membrane.
Mistargeted Mitochondrial Proteins Activate a Proteostatic Response in the Cytosol. Wrobel, Lidia; Topf, Ulrike; Bragoszewski, Piotr; Wiese, Sebastian; Sztolsztener, Malgorzata E.; Oeljeklaus, Silke; Varabyova, Aksana; Lirski, Maciej; Chroscicki, Piotr; Mroczek, Seweryn; Januszewicz, Elzbieta; Dziembowski, Andrzej; Koblowska, Marta; Warscheid, Bettina; Chacinska, Agnieszka (2015). 524(7566) 485–488.
Most of the mitochondrial proteome originates from nuclear genes and is transported into the mitochondria after synthesis in the cytosol. Complex machineries which maintain the specificity of protein import and sorting include the TIM23 translocase responsible for the transfer of precursor proteins into the matrix, and the mitochondrial intermembrane space import and assembly (MIA) machinery required for the biogenesis of intermembrane space proteins. Dysfunction of mitochondrial protein sorting pathways results in diminishing specific substrate proteins, followed by systemic pathology of the organelle and organismal death. The cellular responses caused by accumulation of mitochondrial precursor proteins in the cytosol are mainly unknown. Here we present a comprehensive picture of the changes in the cellular transcriptome and proteome in response to a mitochondrial import defect and precursor over-accumulation stress. Pathways were identified that protect the cell against mitochondrial biogenesis defects by inhibiting protein synthesis and by activation of the proteasome, a major machine for cellular protein clearance. Proteasomal activity is modulated in proportion to the quantity of mislocalized mitochondrial precursor proteins in the cytosol. We propose that this type of unfolded protein response activated by mistargeting of proteins (UPRam) is beneficial for the cells. UPRam provides a means for buffering the consequences of physiological slowdown in mitochondrial protein import and for counteracting pathologies that are caused or contributed by mitochondrial dysfunction.
Assembly of \($\beta$\)-Barrel Proteins in the Mitochondrial Outer Membrane. Höhr, Alexandra I. C.; Straub, Sebastian P.; Warscheid, Bettina; Becker, Thomas; Wiedemann, Nils (2015). 1853(1) 74–88.
Mitochondria evolved through endosymbiosis of a Gram-negative progenitor with a host cell to generate eukaryotes. Therefore, the outer membrane of mitochondria and Gram-negative bacteria contain pore proteins with \($\beta$\)-barrel topology. After synthesis in the cytosol, \($\beta$\)-barrel precursor proteins are first transported into the mitochondrial intermembrane space. Folding and membrane integration of \($\beta$\)-barrel proteins depend on the mitochondrial sorting and assembly machinery (SAM) located in the outer membrane, which is related to the \($\beta$\)-barrel assembly machinery (BAM) in bacteria. The SAM complex recognizes \($\beta$\)-barrel proteins by a \($\beta$\)-signal in the C-terminal \($\beta$\)-strand that is required to initiate \($\beta$\)-barrel protein insertion into the outer membrane. In addition, the SAM complex is crucial to form membrane contacts with the inner mitochondrial membrane by interacting with the mitochondrial contact site and cristae organizing system (MICOS) and shares a subunit with the endoplasmic reticulum-mitochondria encounter structure (ERMES) that links the outer mitochondrial membrane to the endoplasmic reticulum (ER).
Mitochondrial Heat Shock Protein (Hsp) 70 and Hsp10 Cooperate in the Formation of Hsp60 Complexes. Böttinger, Lena; Oeljeklaus, Silke; Guiard, Bernard; Rospert, Sabine; Warscheid, Bettina; Becker, Thomas (2015). 290(18) 11611–11622.
Mitochondrial Hsp70 (mtHsp70) mediates essential functions for mitochondrial biogenesis, like import and folding of proteins. In these processes, the chaperone cooperates with cochaperones, the presequence translocase, and other chaperone systems. The chaperonin Hsp60, together with its cofactor Hsp10, catalyzes folding of a subset of mtHsp70 client proteins. Hsp60 forms heptameric ring structures that provide a cavity for protein folding. How the Hsp60 rings are assembled is poorly understood. In a comprehensive interaction study, we found that mtHsp70 associates with Hsp60 and Hsp10. Surprisingly, mtHsp70 interacts with Hsp10 independently of Hsp60. The mtHsp70-Hsp10 complex binds to the unassembled Hsp60 precursor to promote its assembly into mature Hsp60 complexes. We conclude that coupling to Hsp10 recruits mtHsp70 to mediate the biogenesis of the heptameric Hsp60 rings.
Structural Insights into Cargo Recognition by the Yeast PTS1 Receptor. Hagen, Stefanie; Drepper, Friedel; Fischer, Sven; Fodor, Krisztian; Passon, Daniel; Platta, Harald W.; Zenn, Michael; Schliebs, Wolfgang; Girzalsky, Wolfgang; Wilmanns, Matthias; Warscheid, Bettina; Erdmann, Ralf (2015). 290(44) 26610–26626.
The peroxisomal matrix protein import is facilitated by cycling import receptors that shuttle between the cytosol and the peroxisomal membrane. The import receptor Pex5p mediates the import of proteins harboring a peroxisomal targeting signal of type I (PTS1). Purified recombinant Pex5p forms a dimeric complex with the PTS1-protein Pcs60p in vitro with a KD of 0.19 \($\mu$\)m. To analyze the structural basis for receptor-cargo recognition, the PTS1 and adjacent amino acids of Pcs60p were systematically scanned for Pex5p binding by an in vitro site-directed photo-cross-linking approach. The cross-linked binding regions of the receptor were subsequently identified by high resolution mass spectrometry. Most cross-links were found with TPR6, TPR7, as well as the 7C-loop of Pex5p. Surface plasmon resonance analysis revealed a bivalent interaction mode for Pex5p and Pcs60p. Interestingly, Pcs60p lacking its C-terminal tripeptide sequence was efficiently cross-linked to the same regions of Pex5p. The KD value of the interaction of truncated Pcs60p and Pex5p was in the range of 7.7 \($\mu$\)m. Isothermal titration calorimetry and surface plasmon resonance measurements revealed a monovalent binding mode for the interaction of Pex5p and Pcs60p lacking the PTS1. Our data indicate that Pcs60p contains a second contact site for its receptor Pex5p, beyond the C-terminal tripeptide. The physiological relevance of the ancillary binding region was supported by in vivo import studies. The bivalent binding mode might be explained by a two-step concept as follows: first, cargo recognition and initial tethering by the PTS1-receptor Pex5p; second, lock-in of receptor and cargo.
Phytoene Desaturase from Oryza Sativa: Oligomeric Assembly, Membrane Association and Preliminary 3D-Analysis. Gemmecker, Sandra; Schaub, Patrick; Koschmieder, Julian; Brausemann, Anton; Drepper, Friedel; Rodriguez-Franco, Marta; Ghisla, Sandro; Warscheid, Bettina; Einsle, Oliver; Beyer, Peter (2015). 10(7) e0131717.
Recombinant phytoene desaturase (PDS-His6) from rice was purified to near-homogeneity and shown to be enzymatically active in a biphasic, liposome-based assay system. The protein contains FAD as the sole protein-bound redox-cofactor. Benzoquinones, not replaceable by molecular oxygen, serve as a final electron acceptor defining PDS as a 15-cis-phytoene (donor):plastoquinone oxidoreductase. The herbicidal PDS-inhibitor norflurazon is capable of arresting the reaction by stabilizing the intermediary FAD(red), while an excess of the quinone acceptor relieves this blockage, indicating competition. The enzyme requires its homo-oligomeric association for activity. The sum of data collected through gel permeation chromatography, non-denaturing polyacrylamide electrophoresis, chemical cross-linking, mass spectrometry and electron microscopy techniques indicate that the high-order oligomers formed in solution are the basis for an active preparation. Of these, a tetramer consisting of dimers represents the active unit. This is corroborated by our preliminary X-ray structural analysis that also revealed similarities of the protein fold with the sequence-inhomologous bacterial phytoene desaturase CRTI and other oxidoreductases of the GR2-family of flavoproteins. This points to an evolutionary relatedness of CRTI and PDS yielding different carotene desaturation sequences based on homologous protein folds.
Quantitative Phosphoproteomics Reveals the Protein Tyrosine Kinase Pyk2 as a Central Effector of Olfactory Receptor Signaling in Prostate Cancer Cells. Wiese, Heike; Gelis, Lian; Wiese, Sebastian; Reichenbach, Christa; Jovancevic, Nikolina; Osterloh, Markus; Meyer, Helmut E.; Neuhaus, Eva M.; Hatt, Hanns H.; Radziwill, Gerald; Warscheid, Bettina (2015). 1854(6) 632–640.
The prostate-specific G-protein-coupled receptor 1 (PSGR1) is an olfactory receptor specifically expressed in the prostate gland. PSGR1 expression is elevated both in benign prostatic hyperplasia tissue and in prostate cancer. Stimulation of PSGR1 by the odorant \($\beta$\)-ionone leads to an increase in the intracellular Ca(2+) concentration, activation of mitogen-activated protein (MAP) kinases and a decrease in prostate cancer cell proliferation. To further extend our knowledge about PSGR1 signaling in prostate cancer cells, we performed a quantitative phosphoproteomics study using stable isotope labeling by amino acids in cell culture and mass spectrometry. We report 51 differentially regulated phosphorylation sites in 24 proteins with functions in cytoskeletal remodeling, signaling and ion transport. Activation of PSGR1 evoked an increase in intracellular pH mediated by the sodium/hydrogen exchanger NHE1. Furthermore, we report the protein tyrosine kinase Pyk2 as a central effector of PSGR1 signaling cascades in LNCaP cells. Our data show that phosphorylation of p38 MAP kinase is triggered by Pyk2. In addition, we confirmed dephosphorylation of the tumor suppressor protein N-myc downstream regulated gene 1 (NDRG1) at Ser330 downstream of Pyk2. Since NDRG1 impacts oncogenic signaling pathways interfering with tumor progression, we suggest that the Pyk2-NDRG1 axis is possibly involved in conveying the anti-proliferative effect of \($\beta$\)-ionone in prostate cancer cells. This article is part of a Special Issue entitled: Medical Proteomics.
Role of Membrane Contact Sites in Protein Import into Mitochondria. Horvath, Susanne E.; Rampelt, Heike; Oeljeklaus, Silke; Warscheid, Bettina; van der Laan, Martin; Pfanner, Nikolaus (2015). 24(3) 277–297.
Mitochondria import more than 1,000 different proteins from the cytosol. The proteins are synthesized as precursors on cytosolic ribosomes and are translocated by protein transport machineries of the mitochondrial membranes. Five main pathways for protein import into mitochondria have been identified. Most pathways use the translocase of the outer mitochondrial membrane (TOM) as the entry gate into mitochondria. Depending on specific signals contained in the precursors, the proteins are subsequently transferred to different intramitochondrial translocases. In this article, we discuss the connection between protein import and mitochondrial membrane architecture. Mitochondria possess two membranes. It is a long-standing question how contact sites between outer and inner membranes are formed and which role the contact sites play in the translocation of precursor proteins. A major translocation contact site is formed between the TOM complex and the presequence translocase of the inner membrane (TIM23 complex), promoting transfer of presequence-carrying preproteins to the mitochondrial inner membrane and matrix. Recent findings led to the identification of contact sites that involve the mitochondrial contact site and cristae organizing system (MICOS) of the inner membrane. MICOS plays a dual role. It is crucial for maintaining the inner membrane cristae architecture and forms contacts sites to the outer membrane that promote translocation of precursor proteins into the intermembrane space and outer membrane of mitochondria. The view is emerging that the mitochondrial protein translocases do not function as independent units, but are embedded in a network of interactions with machineries that control mitochondrial activity and architecture.
MITRAC7 Acts as a COX1-Specific Chaperone and Reveals a Checkpoint during Cytochrome c Oxidase Assembly. Dennerlein, Sven; Oeljeklaus, Silke; Jans, Daniel; Hellwig, Christin; Bareth, Bettina; Jakobs, Stefan; Deckers, Markus; Warscheid, Bettina; Rehling, Peter (2015). 12(10) 1644–1655.
Cytochrome c oxidase, the terminal enzyme of the respiratory chain, is assembled from mitochondria- and nuclear-encoded subunits. The MITRAC complex represents the central assembly intermediate during this process as it receives imported subunits and regulates mitochondrial translation of COX1 mRNA. The molecular processes that promote and regulate the progression of assembly downstream of MITRAC are still unknown. Here, we identify MITRAC7 as a constituent of a late form of MITRAC and as a COX1-specific chaperone. MITRAC7 is required for cytochrome c oxidase biogenesis. Surprisingly, loss of MITRAC7 or an increase in its amount causes selective cytochrome c oxidase deficiency in human cells. We demonstrate that increased MITRAC7 levels stabilize and trap COX1 in MITRAC, blocking progression in the assembly process. In contrast, MITRAC7 deficiency leads to turnover of newly synthesized COX1. Accordingly, MITRAC7 affects the biogenesis pathway by stabilizing newly synthesized COX1 in assembly intermediates, concomitantly preventing turnover.
Cox17 Protein Is an Auxiliary Factor Involved in the Control of the Mitochondrial Contact Site and Cristae Organizing System. Chojnacka, Magdalena; Gornicka, Agnieszka; Oeljeklaus, Silke; Warscheid, Bettina; Chacinska, Agnieszka (2015). 290(24) 15304–15312.
The mitochondrial contact site and cristae organizing system (MICOS) is a recently discovered protein complex that is crucial for establishing and maintaining the proper inner membrane architecture and contacts with the outer membrane of mitochondria. The ways in which the MICOS complex is assembled and its integrity is regulated remain elusive. Here, we report a direct link between Cox17, a protein involved in the assembly of cytochrome c oxidase, and the MICOS complex. Cox17 interacts with Mic60, thereby modulating MICOS complex integrity. This interaction does not involve Sco1, a partner of Cox17 in transferring copper ions to cytochrome c oxidase. However, the Cox17-MICOS interaction is regulated by copper ions. We propose that Cox17 is a newly identified factor involved in maintaining the architecture of the MICOS complex.
Mitochondrial Protein Import Receptors in Kinetoplastids Reveal Convergent Evolution over Large Phylogenetic Distances. Mani, Jan; Desy, Silvia; Niemann, Moritz; Chanfon, Astrid; Oeljeklaus, Silke; Pusnik, Mascha; Schmidt, Oliver; Gerbeth, Carolin; Meisinger, Chris; Warscheid, Bettina; Schneider, André (2015). 6 6646.
Mitochondrial protein import is essential for all eukaryotes and mediated by hetero-oligomeric protein translocases thought to be conserved within all eukaryotes. We have identified and analysed the function and architecture of the non-conventional outer membrane (OM) protein translocase in the early diverging eukaryote Trypanosoma brucei. It consists of six subunits that show no obvious homology to translocase components of other species. Two subunits are import receptors that have a unique topology and unique protein domains and thus evolved independently of the prototype receptors Tom20 and Tom70. Our study suggests that protein import receptors were recruited to the core of the OM translocase after the divergence of the major eukaryotic supergroups. Moreover, it links the evolutionary history of mitochondrial protein import receptors to the origin of the eukaryotic supergroups.
Differential Tyrosine Phosphorylation Controls the Function of CNK1 as a Molecular Switch in Signal Transduction. Fischer, Adrian; Brummer, Tilman; Warscheid, Bettina; Radziwill, Gerald (2015). 1853(11 Pt A) 2847–2855.
Scaffold proteins are multidomain proteins without enzymatic function that play a central role in coordinating signaling processes. The scaffold protein CNK1 interacts with pathway-specific signaling proteins and thereby regulates these respective pathways. Here, we revealed tyrosine phosphorylation as a critical regulation mechanism to control the function of CNK1. We identified Tyr 26 as a PDGF-induced and, additionally, Tyr 519 and Tyr 665 as SRC-induced tyrosine phosphorylation sites. Phosphomimetic mutants indicate that phosphorylation of Tyr 519 recruits CNK1 to the nucleus and additional phosphorylation of Tyr 26 enables CNK1 to promote SRE-dependent gene expression. Contrary, mutants preventing tyrosine phosphorylation promote matrix metalloproteinase MMP14 promoter activity. CNK1-driven cell proliferation partially depends on its tyrosine phosphorylation. Upon PDGF stimulation, CNK1 is recruited to the plasma membrane mediated by SRC. Knock down of CNK1 prevents PDGF-induced SRE-dependent gene expression, MMP14 promoter activity and cell proliferation. Thus, tyrosine phosphorylation is an important mechanism to control the subcellular localization of CNK1 and its distinct biological functions.
SILAC Labeling of Yeast for the Study of Membrane Protein Complexes. Oeljeklaus, Silke; Schummer, Andreas; Suppanz, Ida; Warscheid, Bettina (2014). 1188 23–46.
Despite their simplicity compared to multicellular organisms, single-celled yeasts such as the baker's yeast Saccharomyces cerevisiae are widely recognized as model organisms for the study of eukaryotic cell biology. To gain deeper insights into the molecular mechanisms underlying cellular processes, it is of utmost interest to establish the interactome of distinct proteins and to thoroughly analyze the composition of individual protein complexes and their dynamics. Combining affinity purification of epitope-tagged proteins with high-resolution mass spectrometry and quantitative proteomics strategies, in particular stable isotope labeling by amino acids in cell culture (SILAC), represents an unbiased and powerful approach for a most accurate characterization of protein complexes. In this chapter, we provide detailed protocols for the generation of yeast strains (S. cerevisiae) amenable to SILAC-labeling, for epitope tagging of a protein of interest for affinity purification, and for the SILAC-based characterization of membrane protein complexes including the identification of stable core components and transient interaction partners.
The Mitochondrial ADP/ATP Carrier Associates with the Inner Membrane Presequence Translocase in a Stoichiometric Manner. Mehnert, Carola S.; Rampelt, Heike; Gebert, Michael; Oeljeklaus, Silke; Schrempp, Sandra G.; Kochbeck, Lioba; Guiard, Bernard; Warscheid, Bettina; van der Laan, Martin (2014). 289(39) 27352–27362.
The majority of mitochondrial proteins are synthesized with amino-terminal signal sequences. The presequence translocase of the inner membrane (TIM23 complex) mediates the import of these preproteins. The essential TIM23 core complex closely cooperates with partner protein complexes like the presequence translocase-associated import motor and the respiratory chain. The inner mitochondrial membrane also contains a large number of metabolite carriers, but their association with preprotein translocases has been controversial. We performed a comprehensive analysis of the TIM23 interactome based on stable isotope labeling with amino acids in cell culture. Subsequent biochemical studies on identified partner proteins showed that the mitochondrial ADP/ATP carrier associates with the membrane-embedded core of the TIM23 complex in a stoichiometric manner, revealing an unexpected connection of mitochondrial protein biogenesis to metabolite transport. Our data indicate that direct TIM23-AAC coupling may support preprotein import into mitochondria when respiratory activity is low.
The Membrane Proteome of Sensory Cilia to the Depth of Olfactory Receptors. Kuhlmann, Katja; Tschapek, Astrid; Wiese, Heike; Eisenacher, Martin; Meyer, Helmut E.; Hatt, Hanns H.; Oeljeklaus, Silke; Warscheid, Bettina (2014). 13(7) 1828–1843.
In the nasal cavity, the nonmotile cilium of olfactory sensory neurons (OSNs) constitutes the chemosensory interface between the ambient environment and the brain. The unique sensory organelle facilitates odor detection for which it includes all necessary components of initial and downstream olfactory signal transduction. In addition to its function in olfaction, a more universal role in modulating different signaling pathways is implicated, for example, in neurogenesis, apoptosis, and neural regeneration. To further extend our knowledge about this multifunctional signaling organelle, it is of high importance to establish a most detailed proteome map of the ciliary membrane compartment down to the level of transmembrane receptors. We detached cilia from mouse olfactory epithelia via Ca(2+)/K(+) shock followed by the enrichment of ciliary membrane proteins at alkaline pH, and we identified a total of 4,403 proteins by gel-based and gel-free methods in conjunction with high resolution LC/MS. This study is the first to report the detection of 62 native olfactory receptor proteins and to provide evidence for their heterogeneous expression at the protein level. Quantitative data evaluation revealed four ciliary membrane-associated candidate proteins (the annexins ANXA1, ANXA2, ANXA5, and S100A5) with a suggested function in the regulation of olfactory signal transduction, and their presence in ciliary structures was confirmed by immunohistochemistry. Moreover, we corroborated the ciliary localization of the potassium-dependent Na(+)/Ca(2+) exchanger (NCKX) 4 and the plasma membrane Ca(2+)-ATPase 1 (PMCA1) involved in olfactory signal termination, and we detected for the first time NCKX2 in olfactory cilia. Through comparison with transcriptome data specific for mature, ciliated OSNs, we finally delineated the membrane ciliome of OSNs. The membrane proteome of olfactory cilia established here is the most complete today, thus allowing us to pave new avenues for the study of diverse molecular functions and signaling pathways in and out of olfactory cilia and thus to advance our understanding of the biology of sensory organelles in general.
Mitochondrial Translation Factors of Trypanosoma Brucei: Elongation Factor-Tu Has a Unique Subdomain That Is Essential for Its Function. Cristodero, Marina; Mani, Jan; Oeljeklaus, Silke; Aeberhard, Lukas; Hashimi, Hassan; Ramrath, David J. F.; Lukev s, Julius; Warscheid, Bettina; Schneider, André (2013). 90(4) 744–755.
Mitochondrial translation in the parasitic protozoan Trypanosoma brucei relies on imported eukaryotic-type tRNAs as well as on bacterial-type ribosomes that have the shortest known rRNAs. Here we have identified the mitochondrial translation elongation factors EF-Tu, EF-Ts, EF-G1 and release factor RF1 of trypanosomatids and show that their ablation impairs growth and oxidative phosphorylation. In vivo labelling experiments and a SILAC-based analysis of the global proteomic changes induced by EF-Tu RNAi directly link EF-Tu to mitochondrial translation. Moreover, EF-Tu RNAi reveals downregulation of many nuclear encoded subunits of cytochrome oxidase as well as of components of the bc1-complex, whereas most cytosolic ribosomal proteins were upregulated. Interestingly, T.,brucei>EF-Tu has a 30-amino-acid-long, highly charged subdomain, which is unique to trypanosomatids. A combination of RNAi and complementation experiments shows that this subdomain is essential for EF-Tu function, but that it can be replaced by a similar sequence found in eukaryotic EF-1a, the cytosolic counterpart of EF-Tu. A recent cryo-electron microscopy study revealed that trypanosomatid mitochondrial ribosomes have a unique intersubunit space that likely harbours the EF-Tu binding site. These findings suggest that the trypanosomatid-specific EF-Tu subdomain serves as an adaption for binding to these unusual mitochondrial ribosomes.
A Combined Approach of Quantitative Interaction Proteomics and Live-Cell Imaging Reveals a Regulatory Role for Endoplasmic Reticulum (ER) Reticulon Homology Proteins in Peroxisome Biogenesis. David, Christine; Koch, Johannes; Oeljeklaus, Silke; Laernsack, Alexandra; Melchior, Sophie; Wiese, Sebastian; Schummer, Andreas; Erdmann, Ralf; Warscheid, Bettina; Brocard, Cécile (2013). 12(9) 2408–2425.
Peroxisome biogenesis initiates at the endoplasmic reticulum (ER) and maturation allows for the formation of metabolically active organelles. Yet, peroxisomes can also multiply by growth and division. Several proteins, called peroxins, are known to participate in these processes but little is known about their organization to orchestrate peroxisome proliferation. Here, we demonstrate that regulation of peroxisome proliferation relies on the integrity of the tubular ER network. Using a dual track SILAC-based quantitative interaction proteomics approach, we established a comprehensive network of stable as well as transient interactions of the peroxin Pex30p, an integral membrane protein. Through association with merely ER resident proteins, in particular with proteins containing a reticulon homology domain, and with other peroxins, Pex30p designates peroxisome contact sites at ER subdomains. We show that Pex30p traffics through the ER and segregates in punctae to which peroxisomes specifically append, and we ascertain its transient interaction with all subunits of the COPI coatomer complex suggesting the involvement of a vesicle-mediated transport. We establish that the membrane protein Pex30p facilitates the connection of peroxisomes to the ER. Taken together, our data indicate that Pex30p-containing protein complexes act as focal points from which peroxisomes can form and that the tubular ER architecture organized by the reticulon homology proteins Rtn1p, Rtn2p and Yop1p controls this process.
The Proteome of Human Liver Peroxisomes: Identification of Five New Peroxisomal Constituents by a Label-Free Quantitative Proteomics Survey. Gronemeyer, Thomas; Wiese, Sebastian; Ofman, Rob; Bunse, Christian; Pawlas, Magdalena; Hayen, Heiko; Eisenacher, Martin; Stephan, Christian; Meyer, Helmut E.; Waterham, Hans R.; Erdmann, Ralf; Wanders, Ronald J.; Warscheid, Bettina (2013). 8(2) e57395.
The peroxisome is a key organelle of low abundance that fulfils various functions essential for human cell metabolism. Severe genetic diseases in humans are caused by defects in peroxisome biogenesis or deficiencies in the function of single peroxisomal proteins. To improve our knowledge of this important cellular structure, we studied for the first time human liver peroxisomes by quantitative proteomics. Peroxisomes were isolated by differential and Nycodenz density gradient centrifugation. A label-free quantitative study of 314 proteins across the density gradient was accomplished using high resolution mass spectrometry. By pairing statistical data evaluation, cDNA cloning and in vivo colocalization studies, we report the association of five new proteins with human liver peroxisomes. Among these, isochorismatase domain containing 1 protein points to the existence of a new metabolic pathway and hydroxysteroid dehydrogenase like 2 protein is likely involved in the transport or \($\beta$\)-oxidation of fatty acids in human peroxisomes. The detection of alcohol dehydrogenase 1A suggests the presence of an alternative alcohol-oxidizing system in hepatic peroxisomes. In addition, lactate dehydrogenase A and malate dehydrogenase 1 partially associate with human liver peroxisomes and enzyme activity profiles support the idea that NAD(+) becomes regenerated during fatty acid \($\beta$\)-oxidation by alternative shuttling processes in human peroxisomes involving lactate dehydrogenase and/or malate dehydrogenase. Taken together, our data represent a valuable resource for future studies of peroxisome biochemistry that will advance research of human peroxisomes in health and disease.
A Bacterial Toxin Catalyzing Tyrosine Glycosylation of Rho and Deamidation of Gq and Gi Proteins. Jank, Thomas; Bogdanovi’c, Xenia; Wirth, Christophe; Haaf, Erik; Spoerner, Michael; Böhmer, Kira E.; Steinemann, Marcus; Orth, Joachim H. C.; Kalbitzer, Hans Robert; Warscheid, Bettina; Hunte, Carola; Aktories, Klaus (2013). 20(11) 1273–1280.
Entomopathogenic Photorhabdus asymbiotica is an emerging pathogen in humans. Here, we identified a P. asymbiotica protein toxin (PaTox), which contains a glycosyltransferase and a deamidase domain. PaTox mono-O-glycosylates Y32 (or Y34) of eukaryotic Rho GTPases by using UDP-N-acetylglucosamine (UDP-GlcNAc). Tyrosine glycosylation inhibits Rho activation and prevents interaction with downstream effectors, resulting in actin disassembly, inhibition of phagocytosis and toxicity toward insects and mammalian cells. The crystal structure of the PaTox glycosyltransferase domain in complex with UDP-GlcNAc determined at 1.8-AA resolution represents a canonical GT-A fold and is the smallest glycosyltransferase toxin known. (1)H-NMR analysis identifies PaTox as a retaining glycosyltransferase. The glutamine-deamidase domain of PaTox blocks GTP hydrolysis of heterotrimeric G\($\alpha$\)q/11 and G\($\alpha$\)i proteins, thereby activating RhoA. Thus, PaTox hijacks host GTPase signaling in a bidirectional manner by deamidation-induced activation and glycosylation-induced inactivation of GTPases.
Coupling of Mitochondrial Import and Export Translocases by Receptor-Mediated Supercomplex Formation. Qiu, Jian; Wenz, Lena-Sophie; Zerbes, Ralf M.; Oeljeklaus, Silke; Bohnert, Maria; Stroud, David A.; Wirth, Christophe; Ellenrieder, Lars; Thornton, Nicolas; Kutik, Stephan; Wiese, Sebastian; Schulze-Specking, Agnes; Zufall, Nicole; Chacinska, Agnieszka; Guiard, Bernard; Hunte, Carola; Warscheid, Bettina; van der Laan, Martin; Pfanner, Nikolaus; Wiedemann, Nils; Becker, Thomas (2013). 154(3) 596–608.
The mitochondrial outer membrane harbors two protein translocases that are essential for cell viability: the translocase of the outer mitochondrial membrane (TOM) and the sorting and assembly machinery (SAM). The precursors of \($\beta$\)-barrel proteins use both translocases-TOM for import to the intermembrane space and SAM for export into the outer membrane. It is unknown if the translocases cooperate and where the \($\beta$\)-barrel of newly imported proteins is formed. We established a position-specific assay for monitoring \($\beta$\)-barrel formation in vivo and in organello and demonstrated that the \($\beta$\)-barrel was formed and membrane inserted while the precursor was bound to SAM. \($\beta$\)-barrel formation was inhibited by SAM mutants and, unexpectedly, by mutants of the central import receptor, Tom22. We show that the cytosolic domain of Tom22 links TOM and SAM into a supercomplex, facilitating precursor transfer on the intermembrane space side. Our study reveals receptor-mediated coupling of import and export translocases as a means of precursor channeling.
Promiscuous Targeting of Polytopic Membrane Proteins to SecYEG or YidC by the Escherichia Coli Signal Recognition Particle. Welte, Thomas; Kudva, Renuka; Kuhn, Patrick; Sturm, Lukas; Braig, David; Müller, Matthias; Warscheid, Bettina; Drepper, Friedel; Koch, Hans-Georg (2012). 23(3) 464–479.
Protein insertion into the bacterial inner membrane is facilitated by SecYEG or YidC. Although SecYEG most likely constitutes the major integration site, small membrane proteins have been shown to integrate via YidC. We show that YidC can also integrate multispanning membrane proteins such as mannitol permease or TatC, which had been considered to be exclusively integrated by SecYEG. Only SecA-dependent multispanning membrane proteins strictly require SecYEG for integration, which suggests that SecA can only interact with the SecYEG translocon, but not with the YidC insertase. Targeting of multispanning membrane proteins to YidC is mediated by signal recognition particle (SRP), and we show by site-directed cross-linking that the C-terminus of YidC is in contact with SRP, the SRP receptor, and ribosomal proteins. These findings indicate that SRP recognizes membrane proteins independent of the downstream integration site and that many membrane proteins can probably use either SecYEG or YidC for integration. Because protein synthesis is much slower than protein transport, the use of YidC as an additional integration site for multispanning membrane proteins may prevent a situation in which the majority of SecYEG complexes are occupied by translating ribosomes during cotranslational insertion, impeding the translocation of secretory proteins.
Role of Mitochondrial Inner Membrane Organizing System in Protein Biogenesis of the Mitochondrial Outer Membrane. Bohnert, Maria; Wenz, Lena-Sophie; Zerbes, Ralf M.; Horvath, Susanne E.; Stroud, David A.; von der Malsburg, Karina; Müller, Judith M.; Oeljeklaus, Silke; Perschil, Inge; Warscheid, Bettina; Chacinska, Agnieszka; Veenhuis, Marten; van der Klei, Ida J.; Daum, Günther; Wiedemann, Nils; Becker, Thomas; Pfanner, Nikolaus; van der Laan, Martin (2012). 23(20) 3948–3956.
Mitochondria contain two membranes, the outer membrane and the inner membrane with folded cristae. The mitochondrial inner membrane organizing system (MINOS) is a large protein complex required for maintaining inner membrane architecture. MINOS interacts with both preprotein transport machineries of the outer membrane, the translocase of the outer membrane (TOM) and the sorting and assembly machinery (SAM). It is unknown, however, whether MINOS plays a role in the biogenesis of outer membrane proteins. We have dissected the interaction of MINOS with TOM and SAM and report that MINOS binds to both translocases independently. MINOS binds to the SAM complex via the conserved polypeptide transport-associated domain of Sam50. Mitochondria lacking mitofilin, the large core subunit of MINOS, are impaired in the biogenesis of \($\beta$\)-barrel proteins of the outer membrane, whereas mutant mitochondria lacking any of the other five MINOS subunits import \($\beta$\)-barrel proteins in a manner similar to wild-type mitochondria. We show that mitofilin is required at an early stage of \($\beta$\)-barrel biogenesis that includes the initial translocation through the TOM complex. We conclude that MINOS interacts with TOM and SAM independently and that the core subunit mitofilin is involved in biogenesis of outer membrane \($\beta$\)-barrel proteins.
A Universally Conserved ATPase Regulates the Oxidative Stress Response in Escherichia Coli. Wenk, Meike; Ba, Qiaorui; Erichsen, Veronika; MacInnes, Katherine; Wiese, Heike; Warscheid, Bettina; Koch, Hans-Georg (2012). 287(52) 43585–43598.
YchF is an evolutionarily conserved ATPase of unknown function. In humans, the YchF homologue hOla1 appears to influence cell proliferation and was found to be up-regulated in many tumors. A possible involvement in regulating the oxidative stress response was also suggested, but details on the underlying mechanism are lacking. For gaining insight into YchF function, we used Escherichia coli as a model organism and found that YchF overexpression resulted in H(2)O(2) hypersensitivity. This was not caused by transcriptional or translational down-regulation of H(2)O(2)-scavenging enzymes. Instead, we observed YchF-dependent inhibition of catalase activity and a direct interaction with the major E. coli catalase KatG. KatG inhibition was dependent on the ATPase activity of YchF and was regulated by post-translational modifications, most likely including an H(2)O(2)-dependent dephosphorylation. We furthermore showed that YchF expression is repressed by the transcription factor OxyR and further post-translationally modified in response to H(2)O(2). In summary, our data show that YchF functions as a novel negative regulator of the oxidative stress response in E. coli. Considering the available data on hOla1, YchF/Ola1 most likely execute similar functions in bacteria and humans, and their up-regulation inhibits the ability of the cells to scavenge damaging reactive oxygen species.
Find Pairs: The Module for Protein Quantification of the PeakQuant Software Suite. Eisenacher, Martin; Kohl, Michael; Wiese, Sebastian; Hebeler, Romano; Meyer, Helmut E.; Warscheid, Bettina; Stephan, Christian (2012). 16(9) 457–467.
Accurate quantification of proteins is one of the major tasks in current proteomics research. To address this issue, a wide range of stable isotope labeling techniques have been developed, allowing one to quantitatively study thousands of proteins by means of mass spectrometry. In this article, the FindPairs module of the PeakQuant software suite is detailed. It facilitates the automatic determination of protein abundance ratios based on the automated analysis of stable isotope-coded mass spectrometric data. Furthermore, it implements statistical methods to determine outliers due to biological as well as technical variance of proteome data obtained in replicate experiments. This provides an important means to evaluate the significance in obtained protein expression data. For demonstrating the high applicability of FindPairs, we focused on the quantitative analysis of proteome data acquired in (14)N/(15)N labeling experiments. We further provide a comprehensive overview of the features of the FindPairs software, and compare these with existing quantification packages. The software presented here supports a wide range of proteomics applications, allowing one to quantitatively assess data derived from different stable isotope labeling approaches, such as (14)N/(15)N labeling, SILAC, and iTRAQ. The software is publicly available at http://www.medizinisches-proteom-center.de/software and free for academic use.
MITRAC Links Mitochondrial Protein Translocation to Respiratory-Chain Assembly and Translational Regulation. Mick, David U.; Dennerlein, Sven; Wiese, Heike; Reinhold, Robert; Pacheu-Grau, David; Lorenzi, Isotta; Sasarman, Florin; Weraarpachai, Woranontee; Shoubridge, Eric A.; Warscheid, Bettina; Rehling, Peter (2012). 151(7) 1528–1541.
Mitochondrial respiratory-chain complexes assemble from subunits of dual genetic origin assisted by specialized assembly factors. Whereas core subunits are translated on mitochondrial ribosomes, others are imported after cytosolic translation. How imported subunits are ushered to assembly intermediates containing mitochondria-encoded subunits is unresolved. Here, we report a comprehensive dissection of early cytochrome c oxidase assembly intermediates containing proteins required for normal mitochondrial translation and reveal assembly factors promoting biogenesis of human respiratory-chain complexes. We find that TIM21, a subunit of the inner-membrane presequence translocase, is also present in the major assembly intermediates containing newly mitochondria-synthesized and imported respiratory-chain subunits, which we term MITRAC complexes. Human TIM21 is dispensable for protein import but required for integration of early-assembling, presequence-containing subunits into respiratory-chain intermediates. We establish an unexpected molecular link between the TIM23 transport machinery and assembly of respiratory-chain complexes that regulate mitochondrial protein synthesis in response to their assembly state.
Identification of Core Components and Transient Interactors of the Peroxisomal Importomer by Dual-Track Stable Isotope Labeling with Amino Acids in Cell Culture Analysis. Oeljeklaus, Silke; Reinartz, Benedikt S.; Wolf, Janina; Wiese, Sebastian; Tonillo, Jason; Podwojski, Katharina; Kuhlmann, Katja; Stephan, Christian; Meyer, Helmut E.; Schliebs, Wolfgang; Brocard, Cécile; Erdmann, Ralf; Warscheid, Bettina (2012). 11(4) 2567–2580.
The importomer complex plays an essential role in the biogenesis of peroxisomes by mediating the translocation of matrix proteins across the organellar membrane. A central part of this highly dynamic import machinery is the docking complex consisting of Pex14p, Pex13p, and Pex17p that is linked to the RING finger complex (Pex2p, Pex10p, Pex12p) via Pex8p. To gain detailed knowledge on the molecular players governing peroxisomal matrix protein import and, thus, the integrity and functionality of peroxisomes, we aimed at a most comprehensive investigation of stable and transient interaction partners of Pex14p, the central component of the importomer. To this end, we performed a thorough quantitative proteomics study based on epitope tagging of Pex14p combined with dual-track stable isotope labeling with amino acids in cell culture-mass spectrometry (SILAC-MS) analysis of affinity-purified Pex14p complexes and statistics. The results led to the establishment of the so far most extensive Pex14p interactome, comprising 9 core and further 12 transient components. We confirmed virtually all known Pex14p interaction partners including the core constituents of the importomer as well as Pex5p, Pex11p, Pex15p, and Dyn2p. More importantly, we identified new transient interaction partners (Pex25p, Hrr25p, Esl2p, prohibitin) that provide a valuable resource for future investigations on the functionality, dynamics, and regulation of the peroxisomal importomer.
Mgr2 Promotes Coupling of the Mitochondrial Presequence Translocase to Partner Complexes. Gebert, Michael; Schrempp, Sandra G.; Mehnert, Carola S.; Heißwolf, Anna K.; Oeljeklaus, Silke; Ieva, Raffaele; Bohnert, Maria; von der Malsburg, Karina; Wiese, Sebastian; Kleinschroth, Thomas; Hunte, Carola; Meyer, Helmut E.; Haferkamp, Ilka; Guiard, Bernard; Warscheid, Bettina; Pfanner, Nikolaus; van der Laan, Martin (2012). 197(5) 595–604.
Many mitochondrial proteins are synthesized with N-terminal presequences in the cytosol. The presequence translocase of the inner mitochondrial membrane (TIM23) translocates preproteins into and across the membrane and associates with the matrix-localized import motor. The TIM23 complex consists of three core components and Tim21, which interacts with the translocase of the outer membrane (TOM) and the respiratory chain. We have identified a new subunit of the TIM23 complex, the inner membrane protein Mgr2. Mitochondria lacking Mgr2 were deficient in the Tim21-containing sorting form of the TIM23 complex. Mgr2 was required for binding of Tim21 to TIM23(CORE), revealing a binding chain of TIM23(CORE)-Mgr2/Tim21-respiratory chain. Mgr2-deficient yeast cells were defective in growth at elevated temperature, and the mitochondria were impaired in TOM-TIM23 coupling and the import of presequence-carrying preproteins. We conclude that Mgr2 is a coupling factor of the presequence translocase crucial for cell growth at elevated temperature and for efficient protein import.
Dual Function of Sdh3 in the Respiratory Chain and TIM22 Protein Translocase of the Mitochondrial Inner Membrane. Gebert, Natalia; Gebert, Michael; Oeljeklaus, Silke; von der Malsburg, Karina; Stroud, David A.; Kulawiak, Bogusz; Wirth, Christophe; Zahedi, René P.; Dolezal, Pavel; Wiese, Sebastian; Simon, Oliver; Schulze-Specking, Agnes; Truscott, Kaye N.; Sickmann, Albert; Rehling, Peter; Guiard, Bernard; Hunte, Carola; Warscheid, Bettina; van der Laan, Martin; Pfanner, Nikolaus; Wiedemann, Nils (2011). 44(5) 811–818.
The mitochondrial inner membrane harbors the complexes of the respiratory chain and translocase complexes for precursor proteins. We have identified a further subunit of the carrier translocase (TIM22 complex) that surprisingly is identical to subunit 3 of respiratory complex II, succinate dehydrogenase (Sdh3). The membrane-integral protein Sdh3 plays specific functions in electron transfer in complex II. We show by genetic and biochemical approaches that Sdh3 also plays specific functions in the TIM22 complex. Sdh3 forms a subcomplex with Tim18 and is involved in biogenesis and assembly of the membrane-integral subunits of the TIM22 complex. We conclude that the assembly of Sdh3 with different partner proteins, Sdh4 and Tim18, recruits it to two different mitochondrial membrane complexes with functions in bioenergetics and protein biogenesis, respectively.
Ubp15p, a Ubiquitin Hydrolase Associated with the Peroxisomal Export Machinery. Debelyy, Mykhaylo O.; Platta, Harald W.; Saffian, Delia; Hensel, Astrid; Thoms, Sven; Meyer, Helmut E.; Warscheid, Bettina; Girzalsky, Wolfgang; Erdmann, Ralf (2011). 286(32) 28223–28234.
Peroxisomal matrix protein import is facilitated by cycling receptors shuttling between the cytosol and the peroxisomal membrane. One crucial step in this cycle is the ATP-dependent release of the receptors from the peroxisomal membrane. This step is facilitated by the peroxisomal AAA (ATPases associated with various cellular activities) proteins Pex1p and Pex6p with ubiquitination of the receptor being the main signal for its export. Here we report that the AAA complex contains dislocase as well as deubiquitinating activity. Ubp15p, a ubiquitin hydrolase, was identified as a novel constituent of the complex. Ubp15p partially localizes to peroxisomes and is capable of cleaving off ubiquitin moieties from the type I peroxisomal targeting sequence (PTS1) receptor Pex5p. Furthermore, Ubp15p-deficient cells are characterized by a stress-related PTS1 import defect. The results merge into a picture in which removal of ubiquitin from the PTS1 receptor Pex5p is a specific event and might represent a vital step in receptor recycling.
Composition and Topology of the Endoplasmic Reticulum-Mitochondria Encounter Structure. Stroud, David A.; Oeljeklaus, Silke; Wiese, Sebastian; Bohnert, Maria; Lewandrowski, Urs; Sickmann, Albert; Guiard, Bernard; van der Laan, Martin; Warscheid, Bettina; Wiedemann, Nils (2011). 413(4) 743–750.
Eukaryotic cells contain multiple organelles, which are functionally and structurally interconnected. The endoplasmic reticulum-mitochondria encounter structure (ERMES) forms a junction between mitochondria and the endoplasmic reticulum (ER). Four ERMES proteins are known in yeast, the ER-anchored protein Mmm1 and three mitochondria-associated proteins, Mdm10, Mdm12 and Mdm34, with functions related to mitochondrial morphology and protein biogenesis. We mapped the glycosylation sites of ERMES and demonstrate that three asparagine residues in the N-terminal domain of Mmm1 are glycosylated. While the glycosylation is dispensable, the cytosolic C-terminal domain of Mmm1 that connects to the Mdm proteins is required for Mmm1 function. To analyze the composition of ERMES, we determined the subunits by quantitative mass spectrometry. We identified the calcium-binding GTPase Gem1 as a new ERMES subunit, revealing that ERMES is composed of five genuine subunits. Taken together, ERMES represents a platform that integrates components with functions in formation of ER-mitochondria junctions, maintenance of mitochondrial morphology, protein biogenesis and calcium binding.
Genome-Wide Characterization of miR-34a Induced Changes in Protein and mRNA Expression by a Combined Pulsed SILAC and Microarray Analysis. Kaller, Markus; Liffers, Sven-Thorsten; Oeljeklaus, Silke; Kuhlmann, Katja; Röh, Simone; Hoffmann, Reinhard; Warscheid, Bettina; Hermeking, Heiko (2011). 10(8) M111.010462.
The gene encoding the miR-34a microRNA is a transcriptional target of the p53 tumor suppressor protein and subject to epigenetic inactivation in colorectal cancer and numerous other tumor types. Here, we combined pulsed SILAC (pSILAC) and microarray analyses to identify miR-34a-induced changes in protein and mRNA expression. pSILAC allowed to quantify the de novo protein synthesis of 1206 proteins after activation of a conditional miR-34a allele in a colorectal cancer cell line. \($\sim$\)19% of the detected proteins were differentially regulated, with 113 proteins being down- and 115 up-regulated. The proteins with a miR-34a seed-matching-sequence in the 3'-untranslated region (UTR) of the corresponding mRNA showed a clear bias toward translational repression. Proteins involved in DNA replication, e.g. the MCM proteins, and cell proliferation, were over-represented among indirectly down-regulated proteins lacking a miR-34a seed-match. The decrease in de novo protein synthesis of direct miR-34a targets correlated with reduced levels of the corresponding mRNA in most cases, indicating an interdependence of both types of regulation. In addition, 43 mRNAs encoding proteins not detected by pSILAC were down-regulated after miR-34a expression and contained miR-34a seed-matches. The direct regulation of selected miR-34a target-mRNAs was confirmed using reporter assays. Via down-regulation of the proteins encoded by these mRNAs miR-34a presumably inhibits glycolysis (LDHA), WNT-signaling (LEF1), invasion/migration (AXL) and lipid metabolism (ACSL1, ACSL4). Furthermore, miR-34a may activate p53 by inhibiting its acetylation (MTA2, HDAC1) and degradation (YY1). In summary, miR-34a presumably participates in multiple tumor suppressive pathways by directly and indirectly suppressing the expression of numerous, critical proteins.
Dual Role of Mitofilin in Mitochondrial Membrane Organization and Protein Biogenesis. von der Malsburg, Karina; Müller, Judith M.; Bohnert, Maria; Oeljeklaus, Silke; Kwiatkowska, Paulina; Becker, Thomas; Loniewska-Lwowska, Adrianna; Wiese, Sebastian; Rao, Sanjana; Milenkovic, Dusanka; Hutu, Dana P.; Zerbes, Ralf M.; Schulze-Specking, Agnes; Meyer, Helmut E.; Martinou, Jean-Claude; Rospert, Sabine; Rehling, Peter; Meisinger, Chris; Veenhuis, Marten; Warscheid, Bettina; van der Klei, Ida J.; Pfanner, Nikolaus; Chacinska, Agnieszka; van der Laan, Martin (2011). 21(4) 694–707.
The mitochondrial inner membrane consists of two domains, inner boundary membrane and cristae membrane that are connected by crista junctions. Mitofilin/Fcj1 was reported to be involved in formation of crista junctions, however, different views exist on its function and possible partner proteins. We report that mitofilin plays a dual role. Mitofilin is part of a large inner membrane complex, and we identify five partner proteins as constituents of the mitochondrial inner membrane organizing system (MINOS) that is required for keeping cristae membranes connected to the inner boundary membrane. Additionally, mitofilin is coupled to the outer membrane and promotes protein import via the mitochondrial intermembrane space assembly pathway. Our findings indicate that mitofilin is a central component~of MINOS and functions as a multifunctional regulator of mitochondrial architecture and protein biogenesis.
Mitochondrial Preprotein Translocase of Trypanosomatids Has a Bacterial Origin. Pusnik, Mascha; Schmidt, Oliver; Perry, Andrew J.; Oeljeklaus, Silke; Niemann, Moritz; Warscheid, Bettina; Lithgow, Trevor; Meisinger, Chris; Schneider, André (2011). 21(20) 1738–1743.
Mitochondria are found in all eukaryotic cells and derive from a bacterial endosymbiont [1, 2]. The evolution of a protein import system was a prerequisite for the conversion of the endosymbiont into a true organelle. Tom40, the essential component of the protein translocase of the outer membrane, is conserved in mitochondria of almost all eukaryotes but lacks bacterial orthologs [3-6]. It serves as the gateway through which all mitochondrial proteins are imported. The parasitic protozoa Trypanosoma brucei and its relatives do not have a Tom40-like protein, which raises the question of how proteins are imported by their mitochondria [7, 8]. Using a combination of bioinformatics and in vivo and in vitro studies, we have discovered that T. brucei likely employs a different import channel, termed ATOM (archaic translocase of the outer mitochondrial membrane). ATOM mediates the import of nuclear-encoded proteins into mitochondria and is essential for viability of trypanosomes. It is not related to Tom40 but is instead an ortholog of a subgroup of the Omp85 protein superfamily that is involved in membrane translocation and insertion of bacterial outer membrane proteins [9]. This suggests that the protein import channel in trypanosomes is a relic of an archaic protein transport system that was operational in the ancestor of all eukaryotes.
Coa3 and Cox14 Are Essential for Negative Feedback Regulation of COX1 Translation in Mitochondria. Mick, David U.; Vukotic, Milena; Piechura, Heike; Meyer, Helmut E.; Warscheid, Bettina; Deckers, Markus; Rehling, Peter (2010). 191(1) 141–154.
Regulation of eukaryotic cytochrome oxidase assembly occurs at the level of Cox1 translation, its central mitochondria-encoded subunit. Translation of COX1 messenger RNA is coupled to complex assembly in a negative feedback loop: the translational activator Mss51 is thought to be sequestered to assembly intermediates, rendering it incompetent to promote translation. In this study, we identify Coa3 (cytochrome oxidase assembly factor 3; Yjl062w-A), a novel regulator of mitochondrial COX1 translation and cytochrome oxidase assembly. We show that Coa3 and Cox14 form assembly intermediates with newly synthesized Cox1 and are required for Mss51 association with these complexes. Mss51 exists in equilibrium between a latent, translational resting, and a committed, translation-effective, state that are represented as distinct complexes. Coa3 and Cox14 promote formation of the latent state and thus down-regulate COX1 expression. Consequently, lack of Coa3 or Cox14 function traps Mss51 in the committed state and promotes Cox1 synthesis. Our data indicate that Coa1 binding to sequestered Mss51 in complex with Cox14, Coa3, and Cox1 is essential for full inactivation.
RhoA Regulates Peroxisome Association to Microtubules and the Actin Cytoskeleton. Schollenberger, Lukas; Gronemeyer, Thomas; Huber, Christoph M.; Lay, Dorothee; Wiese, Sebastian; Meyer, Helmut E.; Warscheid, Bettina; Saffrich, Rainer; Peränen, Johan; Gorgas, Karin; Just, Wilhelm W. (2010). 5(11) e13886.
The current view of peroxisome inheritance provides for the formation of new peroxisomes by both budding from the endoplasmic reticulum and autonomous division. Here we investigate peroxisome-cytoskeleton interactions and show by proteomics, biochemical and immunofluorescence analyses that actin, non-muscle myosin IIA (NMM IIA), RhoA, Rho kinase II (ROCKII) and Rab8 associate with peroxisomes. Our data provide evidence that (i) RhoA in its inactive state, maintained for example by C. botulinum toxin exoenzyme C3, dissociates from peroxisomes enabling microtubule-based peroxisomal movements and (ii) dominant-active RhoA targets to peroxisomes, uncouples the organelles from microtubules and favors Rho kinase recruitment to peroxisomes. We suggest that ROCKII activates NMM IIA mediating local peroxisomal constrictions. Although our understanding of peroxisome-cytoskeleton interactions is still incomplete, a picture is emerging demonstrating alternate RhoA-dependent association of peroxisomes to the microtubular and actin cytoskeleton. Whereas association of peroxisomes to microtubules clearly serves bidirectional, long-range saltatory movements, peroxisome-acto-myosin interactions may support biogenetic functions balancing peroxisome size, shape, number, and clustering.
Top-down de Novo Protein Sequencing of a 13.6 kDa Camelid Single Heavy Chain Antibody by Matrix-Assisted Laser Desorption Ionization-Time-of-Flight/Time-of-Flight Mass Spectrometry. Resemann, Anja; Wunderlich, Dirk; Rothbauer, Ulrich; Warscheid, Bettina; Leonhardt, Heinrich; Fuchser, Jens; Kuhlmann, Katja; Suckau, Detlev (2010). 82(8) 3283–3292.
The primary structure of a 13.6 kDa single heavy chain camelid antibody (V(H)H) was determined by matrix-assisted laser desorption ionization-time-of-flight/time-of-flight (MALDI-TOF/TOF) top-down sequence analysis. The majority of the sequence was obtained by mass spectrometric de novo sequencing, with the N-terminal 14 amino acid residues being determined using T(3)-sequencing and database interrogation. The determined sequence was confirmed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis of a tryptic digest, which also provided high-energy collisionally induced dissociation (CID) data permitting the clear assignment of 3 of the 14 isobaric Leu/Ile residues. Five of the 11 Leu/Ile ambiguities could be resolved by homology comparisons with known V(H)H sequences. The monoisotopic molecular weight of the V(H)H was determined by ultrahigh-resolution orthogonal electrospray (ESI)-TOF analysis and found to be 13 610.6066 Da, in excellent agreement with the established sequence. To our knowledge, this is the first time that the entire primary structure of a protein with a molecular weight \($>$\)13 kDa has been established by mass spectrometric top-down sequencing.
Kazal-Type Inhibitors in the Stomach of Panstrongylus Megistus (Triatominae, Reduviidae). Meiser, Christian Karl; Piechura, Heike; Werner, Tanja; Dittmeyer-Schäfer, Saskia; Meyer, Helmut E.; Warscheid, Bettina; Schaub, Günter A.; Balczun, Carsten (2010). 40(4) 345–353.
Triatomines inhibit the clotting of ingested blood in the stomach (anterior midgut). After verifying this phenomenon in Panstrongylus megistus using coagulation assays, a full-length cDNA encoding a Kazal-like inhibitor was amplified by PCR. The open reading frame encodes a putative precursor protein of 412 amino acid residues, which was named PmStKaz and contains seven Kazal-like domains forming four Kazal-type inhibitors. A single domain inhibitor and three double-domain inhibitors possess sequence identities of up to 91% to the respective domains of Kazal-type inhibitors from other triatomines. The gene is expressed in the stomach (anterior midgut) but not in the small intestine (posterior midgut), salivary glands or haemocytes. After hydrophobic interaction chromatography of the stomach contents, four fractions (numbers 1-4) inhibited the activity of trypsin, fraction 2 that of subtilisin A, fractions 1, 3 and 4 that of plasmin, and fractions 3 and 4 that of thrombin. After ion exchange chromatography, MALDI-TOF-MS analysis of the intact proteins in fractions 3 and 4 showed diverse masses correlating to PmStKaz IV-V and PmStKaz II-III, respectively. Both proteins seem to be present in several isoforms with variant amino- and carboxy-terminal ends. In reverse zymography of the proteins of the stomach contents after separation by isoelectric focusing and non-reducing SDS-PAGE, much higher concentrations of isoforms of PmStKaz II-III and IV-V were evident than of PmStKaz I and VI-VII.
Uncoupled Responses of Smad4-deficient Cancer Cells to TNFalpha Result in Secretion of Monomeric Laminin-Gamma2. Zboralski, Dirk; Warscheid, Bettina; Klein-Scory, Susanne; Malas, M. Bassel; Becker, Heiko; Böckmann, Miriam; Meyer, Helmut E.; Schmiegel, Wolff; Simon-Assmann, Patricia; Schwarte-Waldhoff, Irmgard (2010). 9(1) 65.
BACKGROUND: Functional loss of the tumor suppressor Smad4 is involved in pancreatic and colorectal carcinogenesis and has been associated with the acquisition of invasiveness. We have previously demonstrated that the heterotrimeric basement membrane protein laminin-332 is a Smad4 target. Namely, Smad4 functions as a positive transcriptional regulator of all three genes encoding laminin-332; its loss is thus implicated in the reduced or discontinuous deposition of the heterotrimeric basement membrane molecule as evident in carcinomas. Uncoupled expression of laminin genes, on the other hand, namely overexpression of the laminin-gamma2 chain is an impressive marker at invasive edges of carcinomas where tumor cells are maximally exposed to signals from stromal cell types like macrophages. As Smad4 is characterized as an integrator of multiple extracellular stimuli in a strongly contextual manner, we asked if loss of Smad4 may also be involved in uncoupled expression of laminin genes in response to altered environmental stimuli. Here, we address Smad4 dependent effects of the prominent inflammatory cytokine TNFalpha on tumor cells. RESULTS: Smad4-reconstituted colon carcinoma cells like adenoma cells respond to TNFalpha with an increased expression of all three chains encoding laminin-332; coincubation with TGFbeta and TNFalpha leads to synergistic induction and to the secretion of large amounts of the heterotrimer. In contrast, in Smad4-deficient cells TNFalpha can induce expression of the gamma2 and beta3 but not the alpha3 chain. Surprisingly, this uncoupled induction of laminin-332 chains in Smad4-negative cells rather than causing intracellular accumulation is followed by the release of gamma2 into the medium, either in a monomeric form or in complexes with as yet unknown proteins. Soluble gamma2 is associated with increased cell migration. CONCLUSIONS: Loss of Smad4 may lead to uncoupled induction of laminin-gamma2 in response to TNFalpha and may therefore represent one of the mechanisms which underlie accumulation of laminin-gamma2 at the invasive margin of a tumor. The finding, that gamma2 is secreted from tumor cells in significant amounts and is associated with increased cell migration may pave the way for further investigation to better understand its functional relevance for tumor progression.
Immunoscreening of the Extracellular Proteome of Colorectal Cancer Cells. Klein-Scory, Susanne; Kübler, Salwa; Diehl, Hanna; Eilert-Micus, Christina; Reinacher-Schick, Anke; Stühler, Kai; Warscheid, Bettina; Meyer, Helmut E.; Schmiegel, Wolff; Schwarte-Waldhoff, Irmgard (2010). 10 70.
BACKGROUND: The release of proteins from tumors can trigger an immune response in cancer patients involving T lymphocytes and B lymphocytes, which results in the generation of antibodies to tumor-derived proteins. Many studies aim to use humoral immune responses, namely autoantibody profiles, directly, as clinical biomarkers. Alternatively, the antibody immune response as an amplification system for tumor associated alterations may be used to indicate putative protein biomarkers with high sensitivity. Aiming at the latter approach we here have implemented an autoantibody profiling strategy which particularly focuses on proteins released by tumor cells in vitro: the so-called secretome. METHODS: For immunoscreening, the extracellular proteome of five colorectal cancer cell lines was resolved on 2D gels, immobilized on PVDF membranes and used for serological screening with individual sera from 21 colorectal cancer patients and 24 healthy controls. All of the signals from each blot were assigned to a master map, and autoantigen candidates were defined based of the pattern of immunoreactivities. The corresponding proteins were isolated from preparative gels, identified by MALDI-MS and/or by nano-HPLC/ESI-MS/MS and exemplarily confirmed by duplex Western blotting combining the human serum samples with antibodies directed against the protein(s) of interest. RESULTS: From 281 secretome proteins stained with autoantibodies in total we first defined the "background patterns" of frequently immunoreactive extracellular proteins in healthy and diseased people. An assignment of these proteins, among them many nominally intracellular proteins, to the subset of exosomal proteins within the secretomes revealed a large overlap. On this basis we defined and consequently confirmed novel biomarker candidates such as the extreme C-terminus of the extracellular matrix protein agrin within the set of cancer-enriched immunoreactivities. CONCLUSIONS: Our findings suggest, first, that autoantibody responses may be due, in large part, to cross-presentation of antigens to the immune system via exosomes, membrane vesicles released by tumor cells and constituting a significant fraction of the secretome. In addition, this immunosecretomics approach has revealed novel biomarker candidates, some of them secretome-specific, and thus serves as a promising complementary tool to the frequently reported immunoproteomic studies for biomarker discovery.
Identification of PEX33, a Novel Component of the Peroxisomal Docking Complex in the Filamentous Fungus Neurospora Crassa. Managadze, David; Würtz, Christian; Wiese, Sebastian; Schneider, Michael; Girzalsky, Wolfgang; Meyer, Helmut E.; Erdmann, Ralf; Warscheid, Bettina; Rottensteiner, Hanspeter (2010). 89(12) 955–964.
The docking complex of peroxisomal matrix protein import is composed of PEX13 and PEX14 in all species analyzed so far, whereas only yeast appears to possess an additional component, PEX17. In this report we isolated PEX14 complexes of Neurospora crassa. Among the complex constituents, one protein designated as PEX33 possessed homology to PEX14 but only in a short N-terminal domain. The PEX14/PEX33 interaction was verified by means of two-hybrid analysis. Moreover, PEX33 was shown to interact with itself and the PTS1-receptor PEX5. Localization studies demonstrated that PEX33 constitutes a glyoxysomal protein. Growth tests of the pex33 deletion strain revealed a defect of this strain in the biogenesis of glyoxysomes and Woronin bodies. As the function of PEX33 was not redundant to that of PEX14, it is a genuine novel peroxin. Based on our experimental data, the function of PEX33 seems to resemble that of yeast PEX17 despite clear structural differences.
Advancements in Plant Proteomics Using Quantitative Mass Spectrometry. Oeljeklaus, Silke; Meyer, Helmut E.; Warscheid, Bettina (2009). 72(3) 545–554.
Due to innovative advancements in quantitative MS technologies, proteomics has evolved from taking mere "snapshots" of distinct proteomes in a defined state to monitoring, for instance, changes in abundance, location and/or posttranslational modification(s) of proteins under various conditions, thereby facilitating the functional characterization of proteins in large scale experiments. In plant biology, MS-based quantitative proteomics strategies utilizing stable isotope labeling or label-free methods for protein quantification have only recently been started to find increasing application to comparative and functional proteomics analyses. This review summarizes latest trends and applications in MS-based quantitative plant proteomics and provides insight into different technologies available. In addition, the studies presented here illustrate the enormous potential of quantitative MS for the analysis of important functional aspects with the emphasis on organellar and phosphoproteomics as well as dynamics and turnover of proteins in plants.
Identification of Proteomic Differences between Squamous Cell Carcinoma of the Lung and Bronchial Epithelium. Poschmann, Gereon; Sitek, Barbara; Sipos, Bence; Ulrich, Anna; Wiese, Sebastian; Stephan, Christian; Warscheid, Bettina; Klöppel, Günter; Vander Borght, Ann; Ramaekers, Frans C. S.; Meyer, Helmut E.; Stühler, Kai (2009). 8(5) 1105–1116.
Proteins that exhibit different expression levels in normal and malignant lung cells are good candidate biomarkers to improve early diagnosis and intervention. We used a quantitative approach and compared the proteome of microdissected cells from normal human bronchial epithelium and squamous cell carcinoma tumors of histopathological grades G2 and G3. DIGE analysis and subsequent MS-based protein identification revealed that 32 non-redundant proteins were differentially regulated between the respective tissue types. These proteins are mainly involved in energy pathways, cell growth or maintenance mechanisms, protein metabolism, and the regulation of DNA and RNA metabolism. The expression of some of these proteins was analyzed by immunohistochemistry using tissue microarrays containing tissue specimen of 55 patients, including normal bronchial epithelium, squamous cell carcinomas, adenocarcinomas, and large cell carcinomas. The results of the immunohistochemical studies correlated with the proteome study data and revealed that particularly HSP47 and a group of cytokeratins (i.e. cytokeratins 6a, 16, and 17) are significantly co-regulated in squamous cell carcinoma. Furthermore cytokeratin 17 showed significantly higher abundance in G2 grade compared with G3 grade squamous cell carcinomas in both the gel-based and the immunohistochemical analysis. Therefore this protein might be used as a marker for stratification between different tumor grades.
Peroxisomal Targeting of PTS2 Pre-Import Complexes in the Yeast Saccharomyces Cerevisiae. Grunau, Silke; Schliebs, Wolfgang; Linnepe, Ruth; Neufeld, Christian; Cizmowski, Christian; Reinartz, Benedikt; Meyer, Helmut E.; Warscheid, Bettina; Girzalsky, Wolfgang; Erdmann, Ralf (2009). 10(4) 451–460.
Posttranslational matrix protein import into peroxisomes uses either one of the two peroxisomal targeting signals (PTS), PTS1 and PTS2. Unlike the PTS1 receptor Pex5p, the PTS2 receptor Pex7p is necessary but not sufficient to target cargo proteins into the peroxisomal matrix and requires coreceptors. Saccharomyces cerevisiae possesses two coreceptors, Pex18p and Pex21p, with a redundant but not a clearly defined function. To gain further insight into the early events of this import pathway, PTS2 pre-import complexes of S. cerevisiae were isolated and characterized by determination of size and protein composition in wild-type and different mutant strains. Mass spectrometric analysis of the cytosolic PTS2 pre-import complex indicates that Fox3p is the only abundant PTS2 protein under oleate growth conditions. Our data strongly suggest that the formation of the ternary cytosolic PTS2 pre-import complex occurs hierarchically. First, Pex7p recognizes cargo proteins through its PTS2 in the cytosol. In a second step, the coreceptor binds to this complex, and finally, this ternary 150 kDa pre-import complex docks at the peroxisomal membrane, where both the PTS1 and the PTS2 import pathways converge. Gel filtration analysis of membrane-bound subcomplexes suggests that Pex13p provides the initial binding partner at the peroxisomal membrane, whereas Pex14p assembles with Pex18p in high-molecular-weight complexes after or during dissociation of the PTS2 receptor.
Detection of Novel Biomarkers of Liver Cirrhosis by Proteomic Analysis. Mölleken, Christian; Sitek, Barbara; Henkel, Corinna; Poschmann, Gereon; Sipos, Bence; Wiese, Sebastian; Warscheid, Bettina; Broelsch, Christoph; Reiser, Markus; Friedman, Scott L.; Tornoe, Ida; Schlosser, Anders; Klöppel, Günter; Schmiegel, Wolff; Meyer, Helmut E.; Holmskov, Uffe; Stühler, Kai (2009). 49(4) 1257–1266.
Hepatic cirrhosis is a life-threatening disease arising from different chronic liver disorders. One major cause for hepatic cirrhosis is chronic hepatitis C. Chronic hepatitis C is characterized by a highly variable clinical course, with at least 20% developing liver cirrhosis within 40 years. Only liver biopsy allows a reliable evaluation of the course of hepatitis C by grading inflammation and staging fibrosis, and thus serum biomarkers for hepatic fibrosis with high sensitivity and specificity are needed. To identify new candidate biomarkers for hepatic fibrosis, we performed a proteomic approach of microdissected cirrhotic septa and liver parenchyma cells. In cirrhotic septa, we detected an increasing expression of cell structure associated proteins, including actin, prolyl 4-hydroxylase, tropomyosin, calponin, transgelin, and human microfibril-associated protein 4 (MFAP-4). Tropomyosin, calponin, and transgelin reflect a contribution of activated stellate cells/myofibroblasts to chronic liver injury. The expression of tropomyosin, transgelin, and MFAP-4, an extracellular matrix associated protein, were further evaluated by immunohistochemistry. Tropomyosin and MFAP-4 demonstrated high serum levels in patients with hepatic cirrhosis of different causes. CONCLUSION: A quantitative analysis of MFAP-4 serum levels in a large number of patients showed MFAP-4 as novel candidate biomarker with high diagnostic accuracy for prediction of nondiseased liver versus cirrhosis [area under receiver operating characteristic curve (AUC) = 0.97, P \($<$\) 0.0001] as well as stage 0 versus stage 4 fibrosis (AUC = 0.84, P \($<$\) 0.0001), and stages 0 to 3 versus stage 4 fibrosis (AUC = 0.76, P \($<$\) 0.0001).
Study of Early Leaf Senescence in Arabidopsis Thaliana by Quantitative Proteomics Using Reciprocal 14N/15N Labeling and Difference Gel Electrophoresis. Hebeler, Romano; Oeljeklaus, Silke; Reidegeld, Kai A.; Eisenacher, Martin; Stephan, Christian; Sitek, Barbara; Stühler, Kai; Meyer, Helmut E.; Sturre, Marcel J. G.; Dijkwel, Paul P.; Warscheid, Bettina (2008). 7(1) 108–120.
Leaf senescence represents the final stage of leaf development and is associated with fundamental changes on the level of the proteome. For the quantitative analysis of changes in protein abundance related to early leaf senescence, we designed an elaborate double and reverse labeling strategy simultaneously employing fluorescent two-dimensional DIGE as well as metabolic (15)N labeling followed by MS. Reciprocal (14)N/(15)N labeling of entire Arabidopsis thaliana plants showed that full incorporation of (15)N into the proteins of the plant did not cause any adverse effects on development and protein expression. A direct comparison of DIGE and (15)N labeling combined with MS showed that results obtained by both quantification methods correlated well for proteins showing low to moderate regulation factors. Nano HPLC/ESI-MS/MS analysis of 21 protein spots that consistently exhibited abundance differences in nine biological replicates based on both DIGE and MS resulted in the identification of 13 distinct proteins and protein subunits that showed significant regulation in Arabidopsis mutant plants displaying advanced leaf senescence. Ribulose 1,5-bisphosphate carboxylase/oxygenase large and three of its four small subunits were found to be down-regulated, which reflects the degradation of the photosynthetic machinery during leaf senescence. Among the proteins showing higher abundance in mutant plants were several members of the glutathione S-transferase family class phi and quinone reductase. Up-regulation of these proteins fits well into the context of leaf senescence since they are generally involved in the protection of plant cells against reactive oxygen species which are increasingly generated by lipid degradation during leaf senescence. With the exception of one glutathione S-transferase isoform, none of these proteins has been linked to leaf senescence before.
Members of the E2D (UbcH5) Family Mediate the Ubiquitination of the Conserved Cysteine of Pex5p, the Peroxisomal Import Receptor. Grou, Cláudia P.; Carvalho, Andreia F.; Pinto, Manuel P.; Wiese, Sebastian; Piechura, Heike; Meyer, Helmut E.; Warscheid, Bettina; Sá-Miranda, Clara; Azevedo, Jorge E. (2008). 283(21) 14190–14197.
According to current models of peroxisomal biogenesis, newly synthesized peroxisomal matrix proteins are transported into the organelle by Pex5p. Pex5p recognizes these proteins in the cytosol, mediates their membrane translocation, and is exported back into the cytosol in an ATP-dependent manner. We have previously shown that export of Pex5p is preceded by (and requires) monoubiquitination of a conserved cysteine residue present at its N terminus. In yeasts, and probably also in plants, ubiquitination of Pex5p is mediated by a specialized ubiquitin-conjugating enzyme, Pex4p. In mammals, the identity of this enzyme has remained unknown for many years. Here, we provide evidence suggesting that E2D1/2/3 (UbcH5a/b/c) are the mammalian functional counterparts of yeast/plant Pex4p. The mechanistic implications of these findings are discussed.
Shy1 Couples Cox1 Translational Regulation to Cytochrome c Oxidase Assembly. Mick, David U.; Wagner, Karina; van der Laan, Martin; Frazier, Ann E.; Perschil, Inge; Pawlas, Magdalena; Meyer, Helmut E.; Warscheid, Bettina; Rehling, Peter (2007). 26(20) 4347–4358.
Cytochrome c oxidase (complex IV) of the respiratory chain is assembled from nuclear and mitochondrially-encoded subunits. Defects in the assembly process lead to severe human disorders such as Leigh syndrome. Shy1 is an assembly factor for complex IV in Saccharomyces cerevisiae and mutations of its human homolog, SURF1, are the most frequent cause for Leigh syndrome. We report that Shy1 promotes complex IV biogenesis through association with different protein modules; Shy1 interacts with Mss51 and Cox14, translational regulators of Cox1. Additionally, Shy1 associates with the subcomplexes of complex IV that are potential assembly intermediates. Formation of these subcomplexes depends on Coa1 (YIL157c), a novel assembly factor that cooperates with Shy1. Moreover, partially assembled forms of complex IV bound to Shy1 and Cox14 can associate with the bc1 complex to form transitional supercomplexes. We suggest that Shy1 links Cox1 translational regulation to complex IV assembly and supercomplex formation.
Psb27, a Cyanobacterial Lipoprotein, Is Involved in the Repair Cycle of Photosystem II. Nowaczyk, Marc M.; Hebeler, Romano; Schlodder, Eberhard; Meyer, Helmut E.; Warscheid, Bettina; Rögner, Matthias (2006). 18(11) 3121–3131.
Photosystem II (PSII) performs one of the key reactions on our planet: the light-driven oxidation of water. This fundamental but very complex process requires PSII to act in a highly coordinated fashion. Despite detailed structural information on the fully assembled PSII complex, the dynamic aspects of formation, processing, turnover, and degradation of PSII with at least 19 subunits and various cofactors are still not fully understood. Transient complexes are especially difficult to characterize due to low abundance, potential heterogeneity, and instability. Here, we show that Psb27 is involved in the assembly of the water-splitting site of PSII and in the turnover of the complex. Psb27 is a bacterial lipoprotein with a specific lipid modification as shown by matrix-assisted laser-desorption ionization time of flight mass spectrometry. The combination of HPLC purification of four different PSII subcomplexes and (15)N pulse label experiments revealed that lipoprotein Psb27 is part of a preassembled PSII subcomplex that represents a distinct intermediate in the repair cycle of PSII.
A Targeted Proteomics Approach to the Rapid Identification of Bacterial Cell Mixtures by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Warscheid, Bettina; Fenselau, Catherine (2004). 4(10) 2877–2892.
A proteomic approach to the rapid identification of bacteria is presented, which relies on the solubilization of a limited number of proteins from intact cells combined with on-probe tryptic digestion. Within 20 min, complete cleavage products of a limited set of bacterial proteins with molecular masses of about 4-125 kDa were obtained by on-probe digestion with immobilized trypsin. Bacterial peptides suitable for unimolecular decomposition analysis were generated within 5 min, and the sequence information obtained allowed identification of abundant proteins, and accordingly, their bacterial sources via searches in the NCBI database. Analysis of fragmentation products was also shown to allow for identification of bacterial peptides identical in mass but differing slightly in amino acid sequence by manual data analysis. In this work, Bacillus subtilis 168, B. globigii, B. sphaericus 14577, B. cereus T, and B. anthracis Sterne were examined, and various cold shock proteins were identified in all species. In addition, DNA-binding, 60 kDa-heat shock, surface-related and other stress-protective proteins were identified in the bacterial cell digests, and species-specific tryptic peptides could be generated from each of the Bacillus species studied. Bacterial peptides could be analyzed with greater sensitivity and mass accuracy than the parent proteins. The applicability of this targeted proteomics approach to the rapid identification of Bacillus species was further established by analyzing binary cell mixtures.
Complete Sequences of Small Acid-Soluble Proteins from Bacillus Globigii. Whiteaker, Jeffrey R.; Warscheid, Bettina; Pribil, Partick; Hathout, Yetrib; Fenselau, Catherine (2004). 39(10) 1113–1121.
Three abundant small acid-soluble proteins (SASPs) from spores of Bacillus globigii were sequenced using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry with post-source decay and nanoelectrospray collision-induced dissociation tandem mass spectrometry. The proteins were extracted from spores with 1 M HCl. Scanning electron micrographs of spores before and after acid extraction show that the spores retain their overall structure but have a shriveled texture following the acid treatment. Extracted SASPs were purified by high-performance liquid chromatography and molecular masses of the SASPs were identified at 7068 (SASP-1), 7332 (SASP-2), and 8889 (gamma-SASP). De novo peptide sequencing was used to determine the protein sequences. The correct ordering of peptide sequences was aided by mapping overlapping enzymatic digests and by comparison with homologous SASPs from Bacillus stearothermophilus. B. globigii is used in many field tests as a surrogate for B. anthracis. Thus complete SASP sequences from B. globigii will facilitate the development of methods for rapid identification of bacteria based on mass spectrometry and the examination of taxonomic relationships between Bacillus species.
MALDI Analysis of Bacilli in Spore Mixtures by Applying a Quadrupole Ion Trap Time-of-Flight Tandem Mass Spectrometer. Warscheid, Bettina; Jackson, Kathryn; Sutton, Chris; Fenselau, Catherine (2003). 75(20) 5608–5617.
A novel ion trap time-of-flight hybrid mass spectrometer (qIT-TOF MS) has been applied for peptide sequencing in proteolytic digests generated from spore mixtures of Bacilli. The method of on-probe solubilization and in situ proteolytic digestion of small, acid-soluble spore proteins has been recently developed in our laboratory, and microorganism identification in less than 20 min was accomplished. In this study, tryptic peptides were generated in situ from complex spore mixtures of B. subtilis 168, B. globigii, B. thuringiensis subs. Kurstaki, and B. cereus T, respectively. MALDI analysis of bacterial peptides generated was performed with an average mass resolving power of 6200 and a mass accuracy of up to 10 ppm using a trap-TOF tandem configuration. Precursor ions of interest were usually selected and stored in the quadrupole ion trap with their complete isotope distribution by choosing a window of +/- 2 Da. Sequence-specific information on isolated protonated peptides was gained via tandem MS experiments with an average mass resolving power of 4450 for product ion analysis, and protein and bacterial sources were identified by database searching.
Direct Quantitative Analysis of Organic Compounds in the Gas and Particle Phase Using a Modified Atmospheric Pressure Chemical Ionization Source in Combination with Ion Trap Mass Spectrometry. Warscheid, Bettina; Kückelmann, Ulrich; Hoffmann, Thorsten (2003). 75(6) 1410–1417.
A slightly modified atmospheric pressure chemical ionization source is employed for direct quantitative analysis of volatile or semivolatile organic compounds in air. The method described here is based on the direct introduction of an analyte in the gas or particle phase, or both, into the ion source of a commercial ion trap mass spectrometer. For quantitation, a standard solution is directly transferred into the vaporizer unit of the ion source via a deactivated fused-silica capillary by using the sheath liquid syringe pump, which is part of the mass spectrometer. The standard addition procedure is conducted by varying the pump rate of a diluted solution of the standard compound in methanol/water. A N2 sheath gas flow is applied for optimal vaporization and mixing with the analyte gas stream. By performing detailed reagent ion monitoring experiments, it is shown that the relative signal intensity of [M + H]+ ions is dependent on the relative humidity of the analyte gas stream as well as the composition and concentration of CI reagent ions. The method is validated by a comparison of the standard addition results with a calibration test gas of known concentration. To demonstrate the potential of atmospheric pressure chemical ionization mass spectrometry as a quantitative analytical technique for on-line investigations, a tropospherically relevant reaction is carried out in a 493-L reaction chamber at atmospheric pressure and 296 K in synthetic air at 50% relative humidity. Finally, the applicability of the technique to rapidly differentiate between analytes in the gas and particle phase is demonstrated.
Bacillus Spore Identification via Proteolytic Peptide Mapping with a Miniaturized MALDI TOF Mass Spectrometer. English, Robert D.; Warscheid, Bettina; Fenselau, Catherine; Cotter, Robert J. (2003). 75(24) 6886–6893.
An approach is tested here as a rapid screening method for Bacillus spore species employing bacterial peptide analysis with a miniaturized MALDI TOF mass spectrometer. A limited set of tryptic peptides was generated in situ following selective solubilization of the small, acid-soluble protein family (SASP) from spore samples on the MALDI sample holder. To facilitate species identification, a compact database was created comprising masses of the tryptic cleavage products generated in silico from all Bacillus and Clostridium SASPs whose sequences are available in public databases. Experimental measurements were matched against the custom-made database, and a published statistical model was then used to evaluate the probability of false identifications.

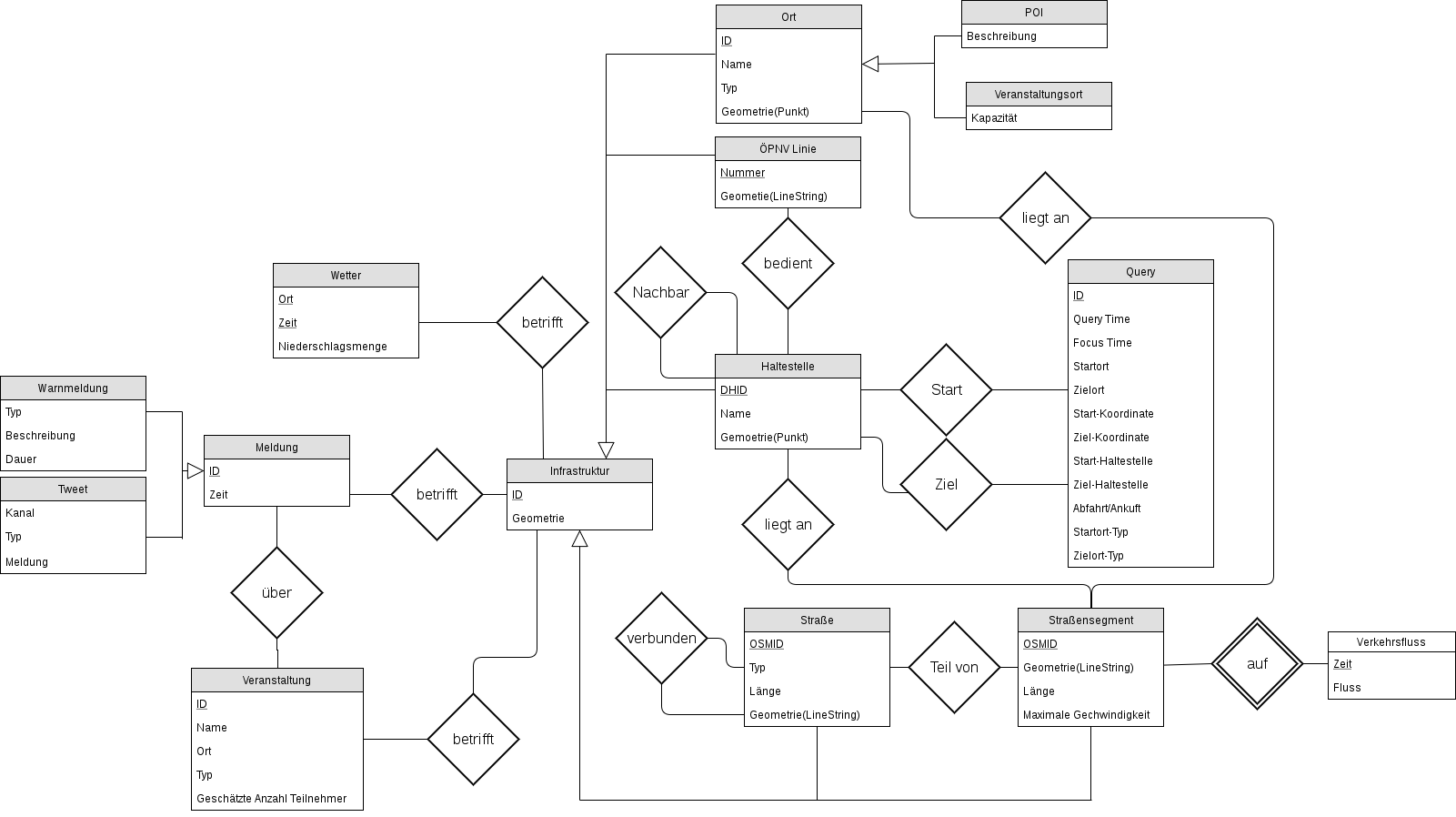{kind=link}